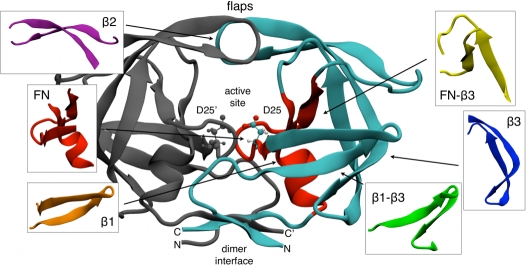
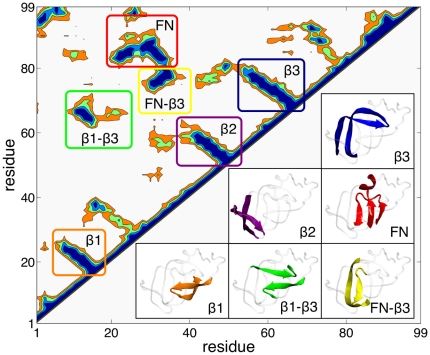
Computational Science, Department of Chemistry and Applied Biosciences, ETH Zurich, Lugano, Switzerland.
PLoS One. 2010 Oct 13;5(10):e13208. doi: 10.1371/journal.pone.0013208.
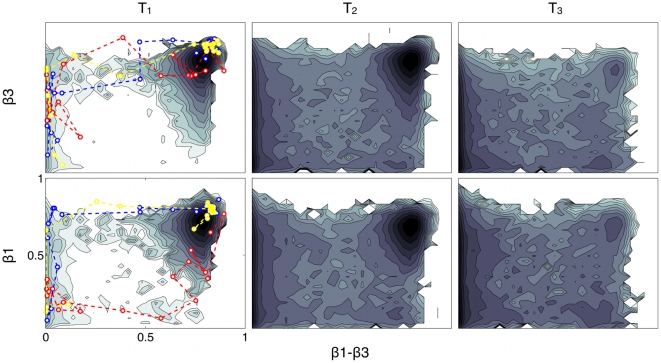
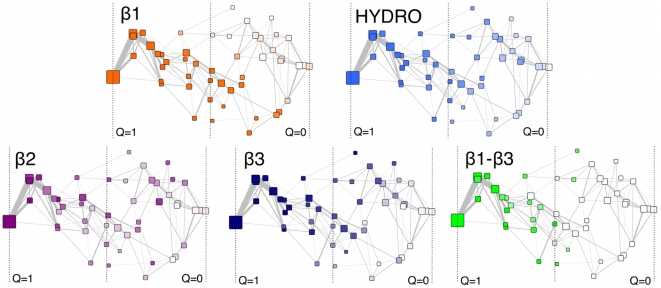
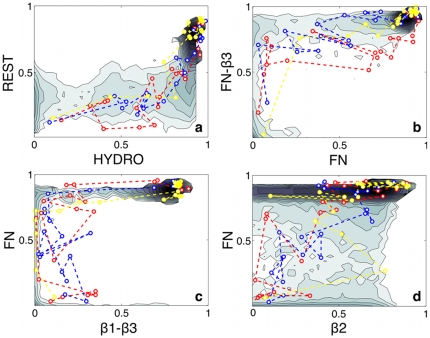
Proteins fold on a time scale incompatible with a mechanism of random search in conformational space thus indicating that somehow they are guided to the native state through a funneled energetic landscape. At the same time the heterogeneous kinetics suggests the existence of several different folding routes. Here we propose a scenario for the folding mechanism of the monomer of HIV-1 protease in which multiple pathways and milestone events coexist. A variety of computational approaches supports this picture. These include very long all-atom molecular dynamics simulations in explicit solvent, an analysis of the network of clusters found in multiple high-temperature unfolding simulations and a complete characterization of free-energy surfaces carried out using a structure-based potential at atomistic resolution and a combination of metadynamics and parallel tempering. Our results confirm that the monomer in solution is stable toward unfolding and show that at least two unfolding pathways exist. In our scenario, the formation of a hydrophobic core is a milestone in the folding process which must occur along all the routes that lead this protein towards its native state. Furthermore, the ensemble of folding pathways proposed here substantiates a rational drug design strategy based on inhibiting the folding of HIV-1 protease.
蛋白质的折叠过程在时间尺度上与构象空间中的随机搜索机制不兼容,这表明它们通过一种被引导到天然状态的能量景观。与此同时,异质动力学表明存在几种不同的折叠途径。在这里,我们提出了一种 HIV-1 蛋白酶单体折叠机制的情景,其中存在多种不同的途径和里程碑事件。各种计算方法都支持这种情况。这些方法包括在明确定义的溶剂中进行非常长的全原子分子动力学模拟,对在多个高温展开模拟中发现的簇网络的分析,以及使用基于结构的势能在原子分辨率下对自由能表面进行的完整特征描述,以及元动力学和并行回火的组合。我们的结果证实,单体在溶液中对展开是稳定的,并表明至少存在两种展开途径。在我们的方案中,形成疏水区是折叠过程中的一个里程碑,它必须沿着所有导致该蛋白质向天然状态的途径发生。此外,这里提出的折叠途径的集合支持了一种基于抑制 HIV-1 蛋白酶折叠的合理药物设计策略。