Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Cambridge, UK.
PLoS Genet. 2010 Oct 14;6(10):e1001154. doi: 10.1371/journal.pgen.1001154.
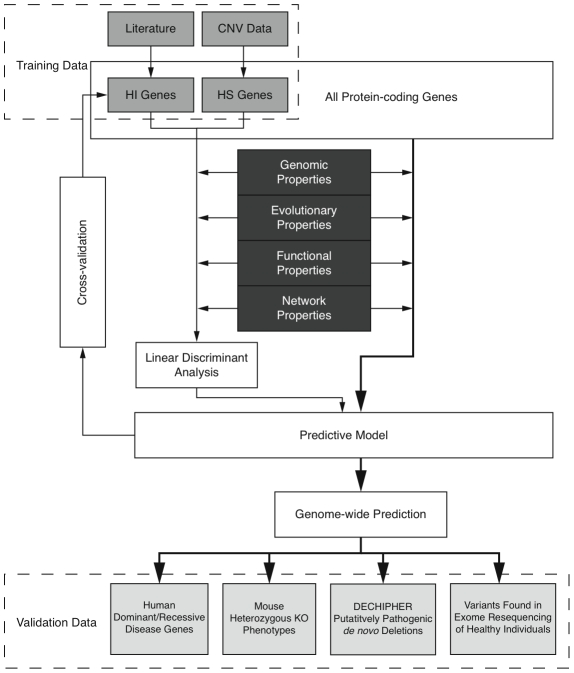
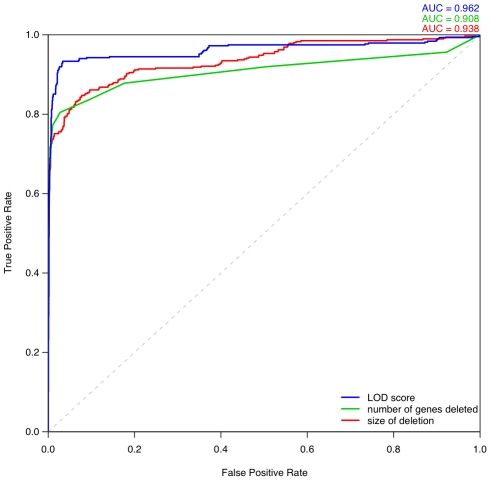
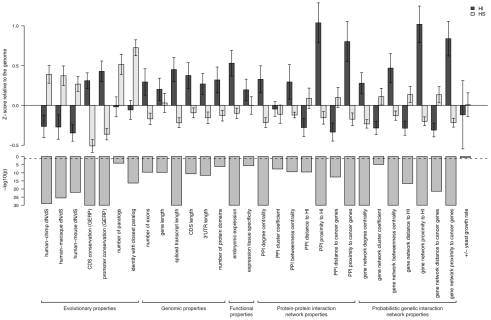
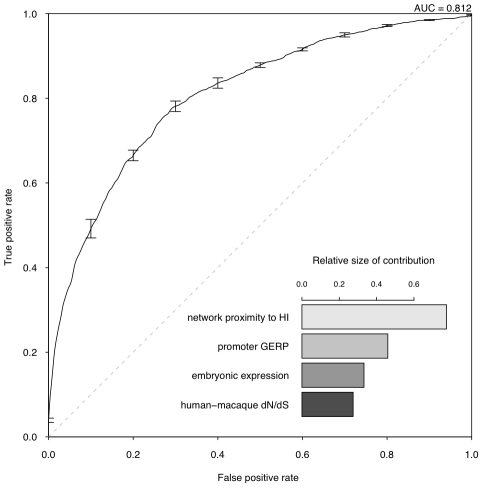
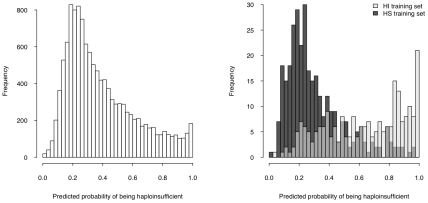
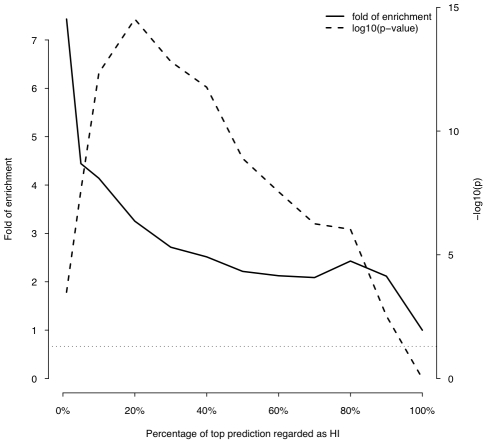
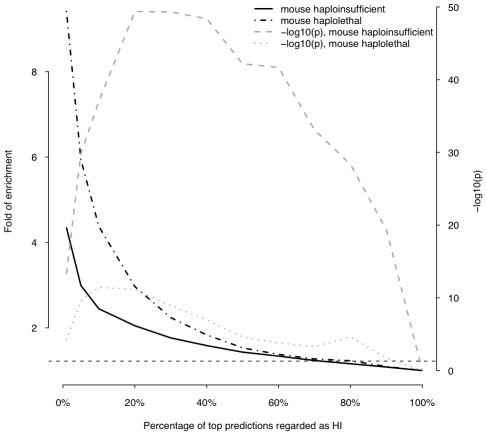
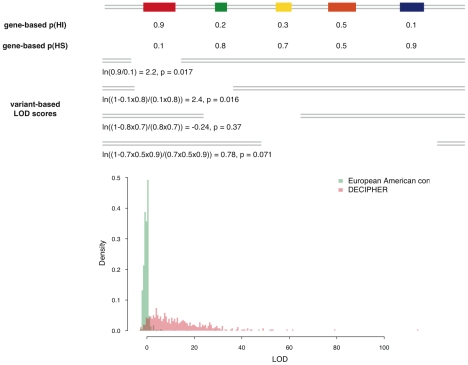
Haploinsufficiency, wherein a single functional copy of a gene is insufficient to maintain normal function, is a major cause of dominant disease. Human disease studies have identified several hundred haploinsufficient (HI) genes. We have compiled a map of 1,079 haplosufficient (HS) genes by systematic identification of genes unambiguously and repeatedly compromised by copy number variation among 8,458 apparently healthy individuals and contrasted the genomic, evolutionary, functional, and network properties between these HS genes and known HI genes. We found that HI genes are typically longer and have more conserved coding sequences and promoters than HS genes. HI genes exhibit higher levels of expression during early development and greater tissue specificity. Moreover, within a probabilistic human functional interaction network HI genes have more interaction partners and greater network proximity to other known HI genes. We built a predictive model on the basis of these differences and annotated 12,443 genes with their predicted probability of being haploinsufficient. We validated these predictions of haploinsufficiency by demonstrating that genes with a high predicted probability of exhibiting haploinsufficiency are enriched among genes implicated in human dominant diseases and among genes causing abnormal phenotypes in heterozygous knockout mice. We have transformed these gene-based haploinsufficiency predictions into haploinsufficiency scores for genic deletions, which we demonstrate to better discriminate between pathogenic and benign deletions than consideration of the deletion size or numbers of genes deleted. These robust predictions of haploinsufficiency support clinical interpretation of novel loss-of-function variants and prioritization of variants and genes for follow-up studies.
单倍体功能不全是指单个基因的功能拷贝不足以维持正常功能,是显性疾病的主要原因。人类疾病研究已经确定了几百个单倍体不足(HI)基因。我们通过系统地识别 8458 名明显健康个体中拷贝数变异明确且反复受损的基因,编制了 1079 个单倍体充足(HS)基因图谱,并对比了这些 HS 基因与已知 HI 基因的基因组、进化、功能和网络特性。我们发现,HI 基因通常更长,编码序列和启动子更保守。HI 基因在早期发育过程中表达水平更高,组织特异性更强。此外,在概率人类功能相互作用网络中,HI 基因具有更多的相互作用伙伴,与其他已知 HI 基因的网络接近度更高。我们基于这些差异建立了一个预测模型,并对 12443 个基因进行了注释,预测它们单倍体不足的概率。我们通过证明具有高预测单倍体不足概率的基因在人类显性疾病相关基因和杂合敲除小鼠中引起异常表型的基因中富集,验证了这些单倍体不足的预测。我们将这些基于基因的单倍体不足预测转化为基因缺失的单倍体不足评分,我们证明与考虑缺失大小或缺失基因数量相比,这些评分能更好地区分致病性和良性缺失。这些稳健的单倍体不足预测支持对新型功能丧失变异的临床解释,并对变体和基因进行优先级排序,以进行后续研究。