Bremel Robert D, Homan E Jane
1ioGenetics LLC, 3591 Anderson Street, Madison, WI 53704, USA.
Immunome Res. 2010 Nov 2;6:8. doi: 10.1186/1745-7580-6-8.
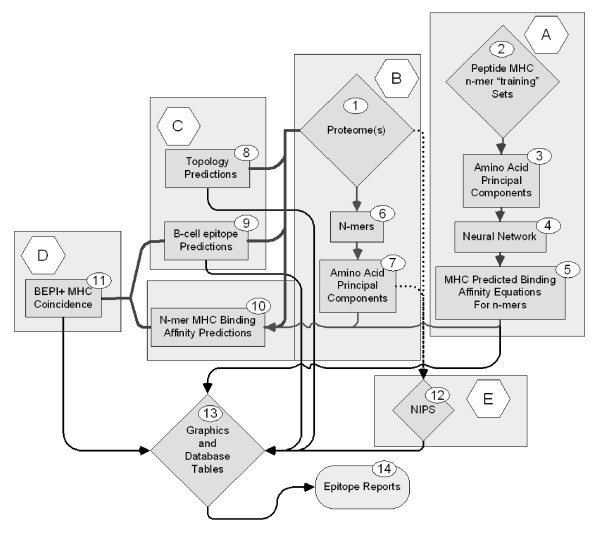
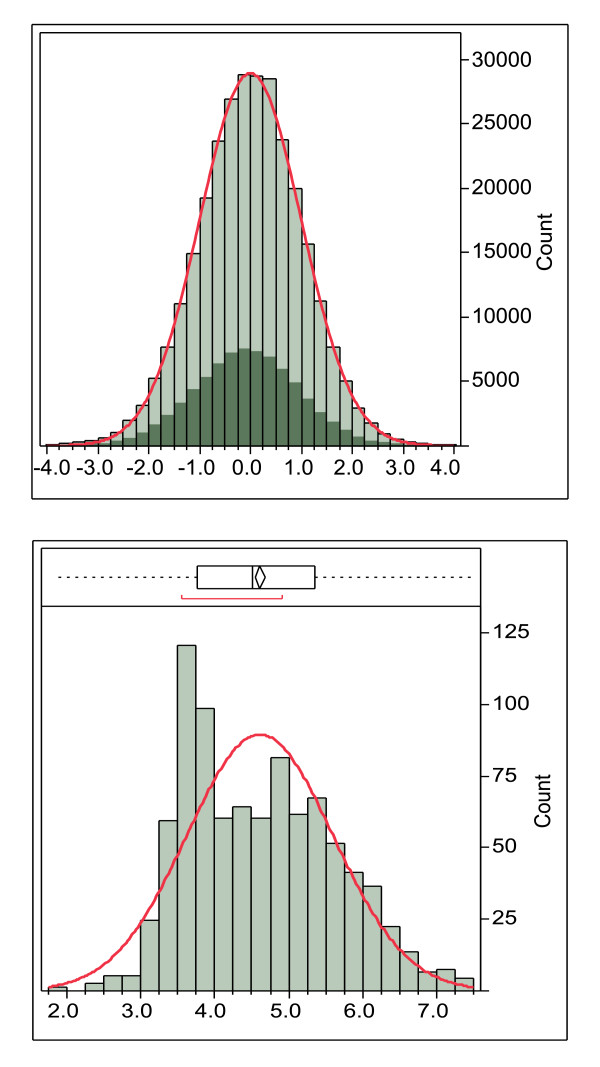
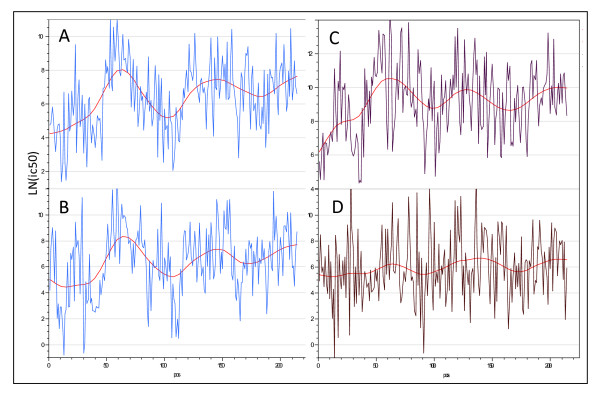
Improving our understanding of the immune response is fundamental to developing strategies to combat a wide range of diseases. We describe an integrated epitope analysis system which is based on principal component analysis of sequences of amino acids, using a multilayer perceptron neural net to conduct QSAR regression predictions for peptide binding affinities to 35 MHC-I and 14 MHC-II alleles.
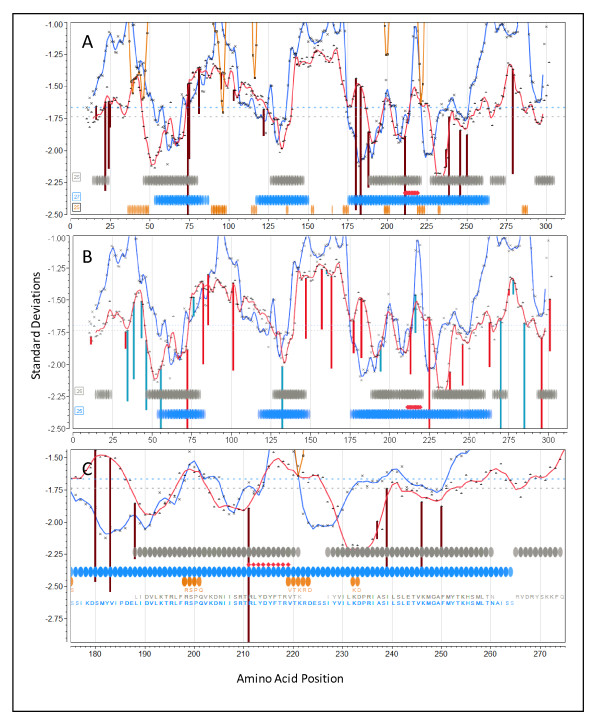
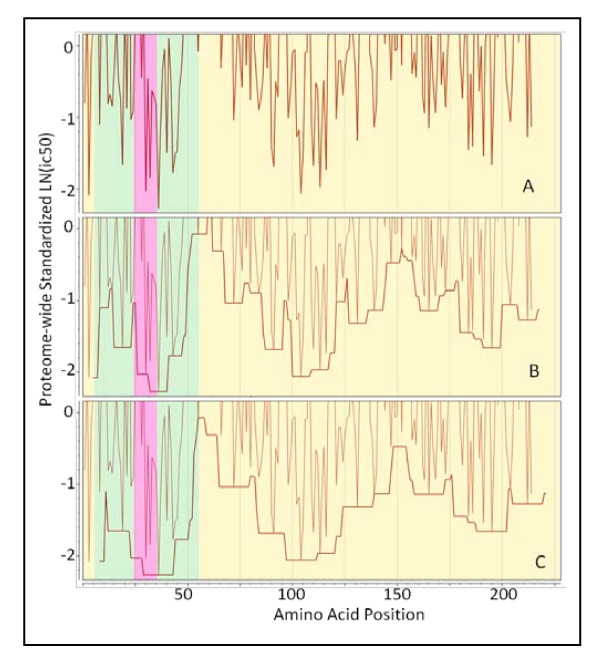
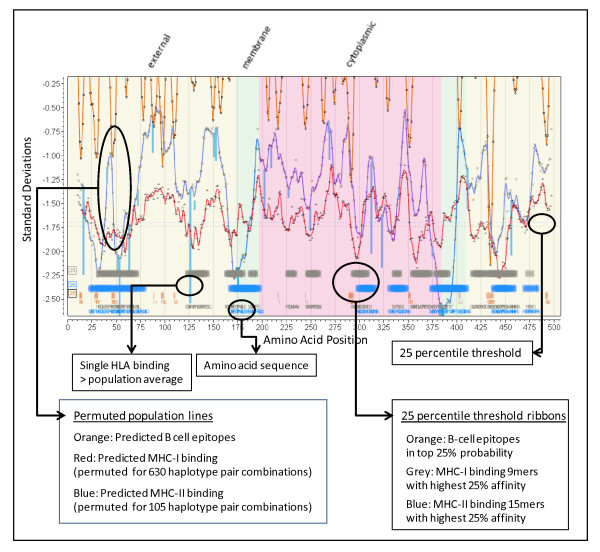
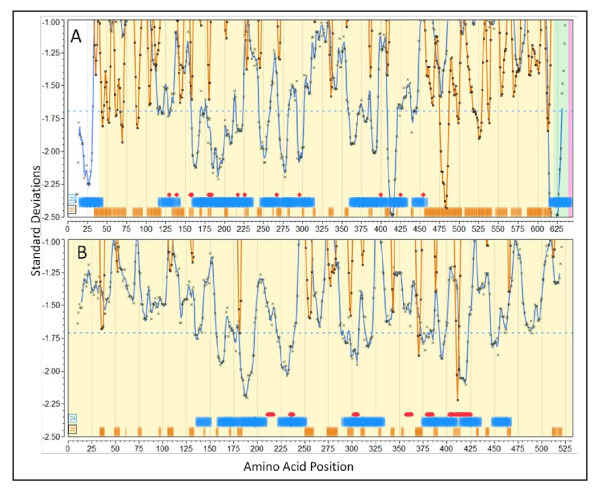
The approach described allows rapid processing of single proteins, entire proteomes or subsets thereof, as well as multiple strains of the same organism. It enables consideration of the interface of diversity of both microorganisms and of host immunogenetics. Patterns of binding affinity are linked to topological features, such as extracellular or intramembrane location, and integrated into a graphical display which facilitates conceptual understanding of the interplay of B-cell and T-cell mediated immunity.Patterns which emerge from application of this approach include the correlations between peptides showing high affinity binding to MHC-I and to MHC-II, and also with predicted B-cell epitopes. These are characterized as coincident epitope groups (CEGs). Also evident are long range patterns across proteins which identify regions of high affinity binding for a permuted population of diverse and heterozygous HLA alleles, as well as subtle differences in reactions with MHCs of individual HLA alleles, which may be important in disease susceptibility, and in vaccine and clinical trial design. Comparisons are shown of predicted epitope mapping derived from application of the QSAR approach with experimentally derived epitope maps from a diverse multi-species dataset, from Staphylococcus aureus, and from vaccinia virus.
A desktop application with interactive graphic capability is shown to be a useful platform for development of prediction and visualization tools for epitope mapping at scales ranging from individual proteins to proteomes from multiple strains of an organism. The possible functional implications of the patterns of peptide epitopes observed are discussed, including their implications for B-cell and T-cell cooperation and cross presentation.
深入了解免疫反应是制定多种疾病防治策略的基础。我们描述了一种综合表位分析系统,该系统基于氨基酸序列的主成分分析,使用多层感知器神经网络对肽与35种MHC-I和14种MHC-II等位基因的结合亲和力进行QSAR回归预测。
所述方法能够快速处理单个蛋白质、整个蛋白质组或其亚组,以及同一生物体的多个菌株。它能够考虑微生物多样性和宿主免疫遗传学的界面。结合亲和力模式与拓扑特征相关联,如细胞外或膜内位置,并整合到图形显示中,便于从概念上理解B细胞和T细胞介导的免疫相互作用。应用该方法出现的模式包括与MHC-I和MHC-II显示高亲和力结合的肽之间的相关性,以及与预测的B细胞表位的相关性。这些被表征为重合表位组(CEG)。跨蛋白质的长程模式也很明显,这些模式识别了针对多种杂合HLA等位基因的置换群体的高亲和力结合区域,以及与个体HLA等位基因的MHC反应的细微差异,这在疾病易感性以及疫苗和临床试验设计中可能很重要。展示了从QSAR方法应用得出的预测表位图谱与来自不同多物种数据集、金黄色葡萄球菌和痘苗病毒的实验得出的表位图谱的比较。
具有交互式图形功能的桌面应用程序被证明是一个有用的平台,可用于开发从单个蛋白质到生物体多个菌株的蛋白质组规模的表位图谱预测和可视化工具。讨论了观察到的肽表位模式可能的功能意义,包括它们对B细胞和T细胞合作及交叉呈递的意义。