Wereszczynski Jeff, McCammon J Andrew
The Department of Chemistry and Biochemistry, University of California, San Diego, 9500 Gilman Drive, La Jolla, California 92093, United States.
J Chem Theory Comput. 2010 Nov 9;6(11):3285-3292. doi: 10.1021/ct100322t. Epub 2010 Oct 13.
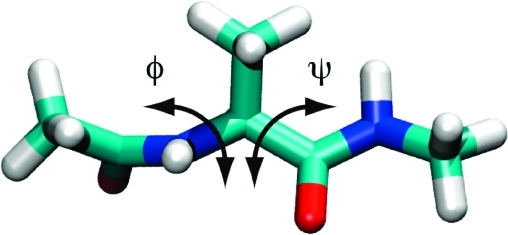
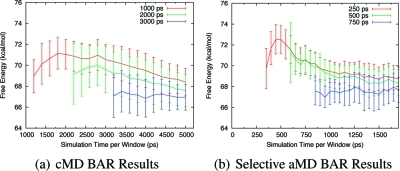
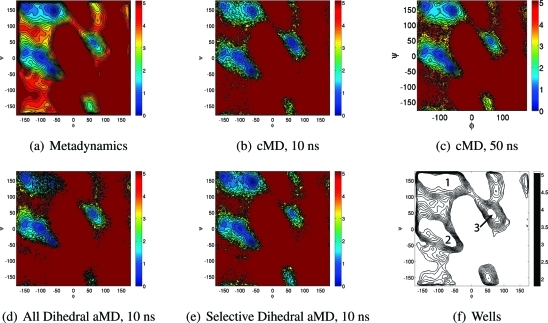
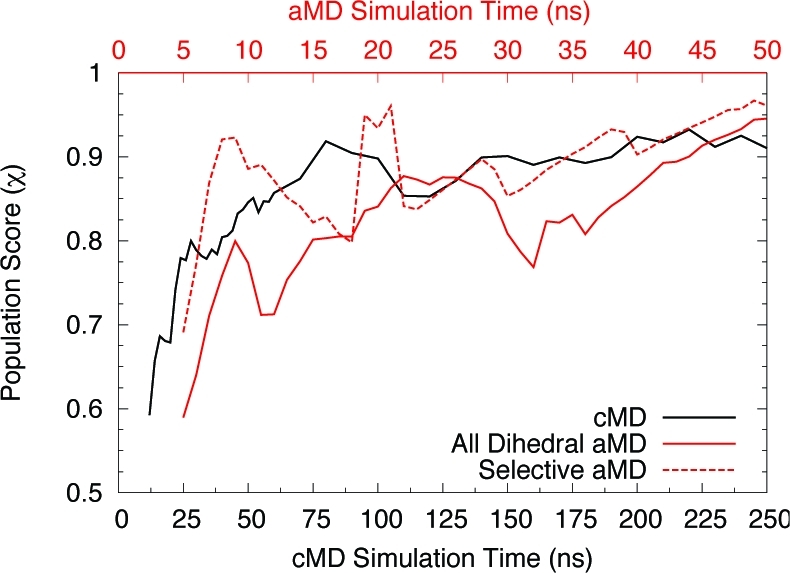
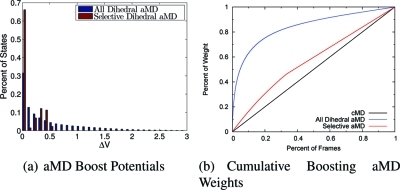
Accelerated molecular dynamics (aMD) has been shown to enhance conformational space sampling relative to classical molecular dynamics; however, the exponential reweighting of aMD trajectories, which is necessary for the calculation of free energies relating to the classical system, is oftentimes problematic, especially for systems larger than small poly peptides. Here, we propose a method of accelerating only the degrees of freedom most pertinent to sampling, thereby reducing the total acceleration added to the system and improving the convergence of calculated ensemble averages, which we term selective aMD. Its application is highlighted in two biomolecular cases. First, the model system alanine dipeptide is simulated with classical MD, all-dihedral aMD, and selective aMD, and these results are compared to the infinite sampling limit as calculated with metadynamics. We show that both forms of aMD enhance the convergence of the underlying free energy landscape by 5-fold relative to classical MD; however, selective aMD can produce improved statistics over all-dihedral aMD due to the improved reweighting. Then we focus on the pharmaceutically relevant case of computing the free energy of the decoupling of oseltamivir in the active site of neuraminidase. Results show that selective aMD greatly reduces the cost of this alchemical free energy transformation, whereas all-dihedral aMD produces unreliable free energy estimates.
与经典分子动力学相比,加速分子动力学(aMD)已被证明能增强构象空间采样;然而,aMD轨迹的指数重加权对于计算与经典系统相关的自由能是必要的,但这一过程常常存在问题,尤其是对于比小多肽更大的系统。在此,我们提出一种方法,仅加速与采样最相关的自由度,从而减少添加到系统的总加速度,并改善计算得到的系综平均值的收敛性,我们将其称为选择性aMD。在两个生物分子案例中突出了其应用。首先,用经典MD、全二面角aMD和选择性aMD对模型系统丙氨酸二肽进行模拟,并将这些结果与用元动力学计算的无限采样极限进行比较。我们表明,相对于经典MD,两种形式的aMD都能将基础自由能景观的收敛性提高5倍;然而,由于重加权得到改善,选择性aMD能比全二面角aMD产生更好的统计结果。然后我们聚焦于计算神经氨酸酶活性位点中奥司他韦解耦自由能这一与药物相关的案例。结果表明,选择性aMD大大降低了这种炼金术自由能转换的成本,而全二面角aMD产生的自由能估计不可靠。