Howard Hughes Medical Institute, Department of Biochemistry and Molecular Biophysics, Columbia University Medical Center, New York, New York, United States of America.
PLoS One. 2010 Nov 8;5(11):e15435. doi: 10.1371/journal.pone.0015435.
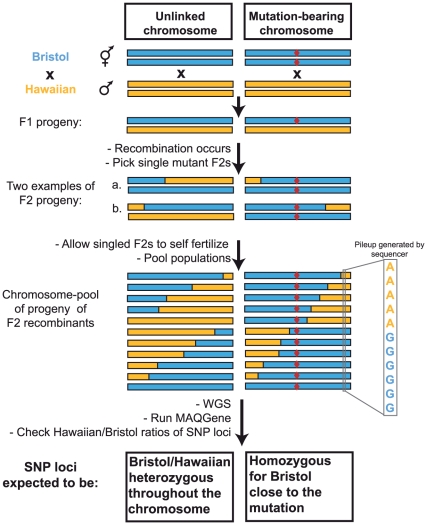
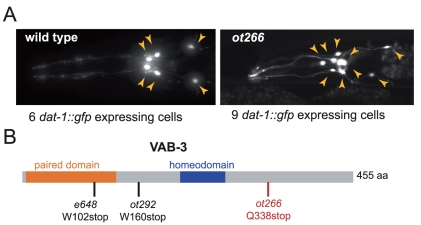
Whole-genome sequencing (WGS) is becoming a fast and cost-effective method to pinpoint molecular lesions in mutagenized genetic model systems, such as Caenorhabditis elegans. As mutagenized strains contain a significant mutational load, it is often still necessary to map mutations to a chromosomal interval to elucidate which of the WGS-identified sequence variants is the phenotype-causing one. We describe here our experience in setting up and testing a simple strategy that incorporates a rapid SNP-based mapping step into the WGS procedure. In this strategy, a mutant retrieved from a genetic screen is crossed with a polymorphic C. elegans strain, individual F2 progeny from this cross is selected for the mutant phenotype, the progeny of these F2 animals are pooled and then whole-genome-sequenced. The density of polymorphic SNP markers is decreased in the region of the phenotype-causing sequence variant and therefore enables its identification in the WGS data. As a proof of principle, we use this strategy to identify the molecular lesion in a mutant strain that produces an excess of dopaminergic neurons. We find that the molecular lesion resides in the Pax-6/Eyeless ortholog vab-3. The strategy described here will further reduce the time between mutant isolation and identification of the molecular lesion.
全基因组测序(WGS)正成为一种快速且经济有效的方法,可精确定位诱变遗传模型系统(如秀丽隐杆线虫)中的分子病变。由于诱变菌株含有大量的突变负荷,因此通常仍需要将突变映射到染色体区间,以阐明 WGS 鉴定的序列变体中哪一个是引起表型的变体。我们在这里描述了我们在建立和测试一种简单策略方面的经验,该策略将快速 SNP 作图步骤纳入 WGS 程序中。在该策略中,从遗传筛选中回收的突变体与多态性秀丽隐杆线虫菌株杂交,从该杂交中选择具有突变表型的单个 F2 后代,将这些 F2 动物的后代混合,然后进行全基因组测序。在引起表型的序列变体区域中,多态 SNP 标记的密度降低,从而能够在 WGS 数据中识别它。作为原理验证,我们使用该策略来鉴定产生过多多巴胺能神经元的突变株中的分子病变。我们发现,分子病变位于 Pax-6/Eyeless 同源物 vab-3 中。本文描述的策略将进一步缩短从突变体分离到鉴定分子病变的时间。