Knowledge Management in Bioinformatics, Humboldt-Universitat zu Berlin Unter den Linden 6, 10099 Berlin, Germany.
BMC Genomics. 2010 Dec 20;11:717. doi: 10.1186/1471-2164-11-717.
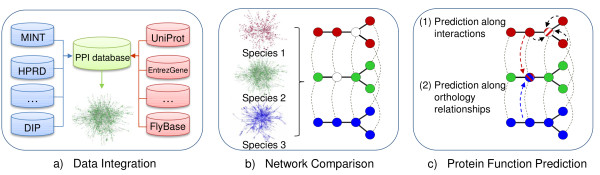
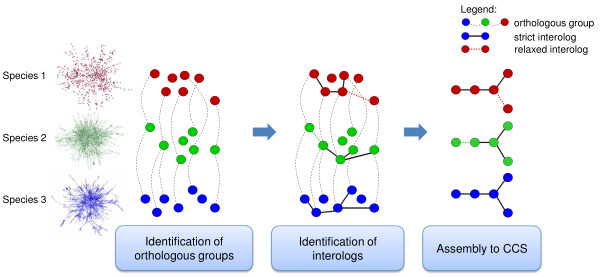
While the number of newly sequenced genomes and genes is constantly increasing, elucidation of their function still is a laborious and time-consuming task. This has led to the development of a wide range of methods for predicting protein functions in silico. We report on a new method that predicts function based on a combination of information about protein interactions, orthology, and the conservation of protein networks in different species.
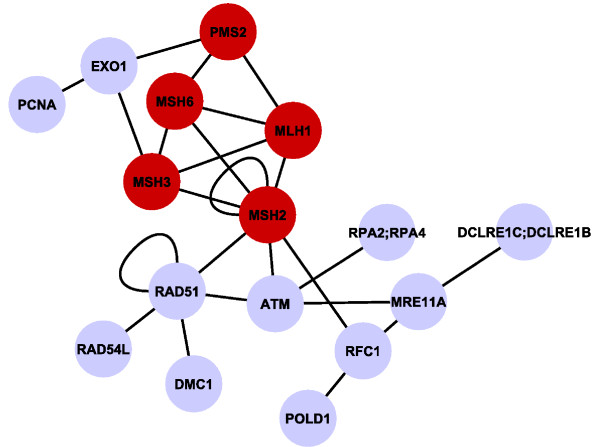
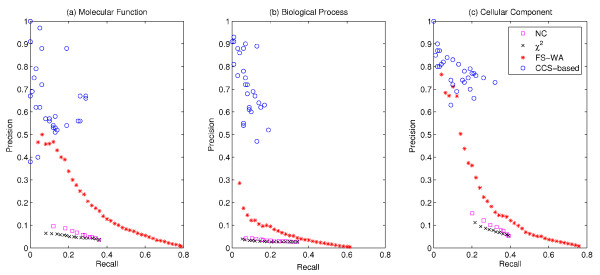
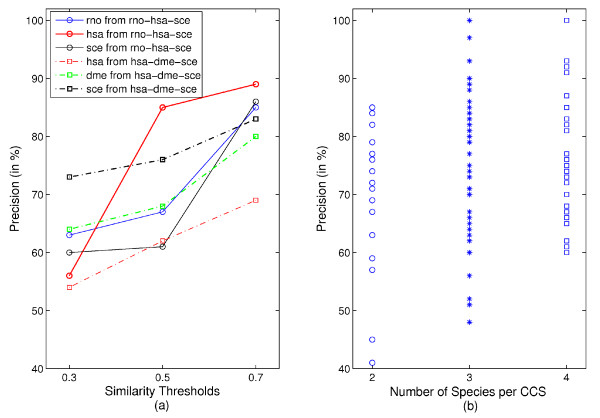
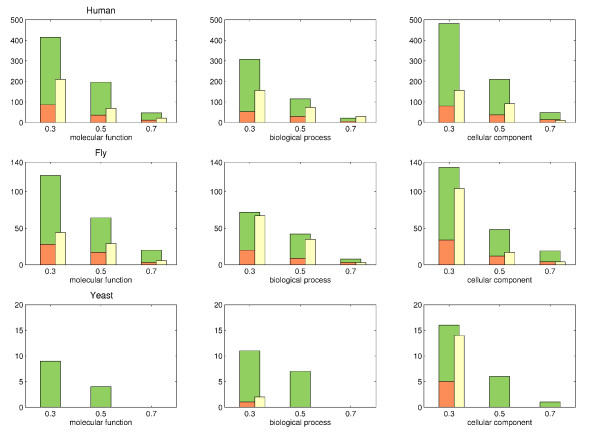
We show that aggregation of these independent sources of evidence leads to a drastic increase in number and quality of predictions when compared to baselines and other methods reported in the literature. For instance, our method generates more than 12,000 novel protein functions for human with an estimated precision of ~76%, among which are 7,500 new functional annotations for 1,973 human proteins that previously had zero or only one function annotated. We also verified our predictions on a set of genes that play an important role in colorectal cancer (MLH1, PMS2, EPHB4 ) and could confirm more than 73% of them based on evidence in the literature.
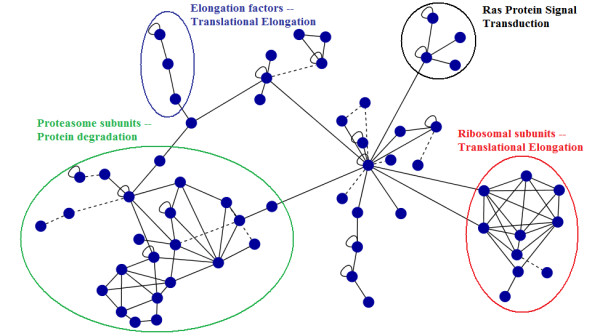
The combination of different methods into a single, comprehensive prediction method infers thousands of protein functions for every species included in the analysis at varying, yet always high levels of precision and very good coverage.
虽然新测序的基因组和基因数量不断增加,但阐明它们的功能仍然是一项费力且耗时的任务。这导致了广泛的方法来预测蛋白质的功能。我们报告了一种新的方法,该方法基于蛋白质相互作用、同源性和不同物种中蛋白质网络的保守性信息的组合来预测功能。
我们表明,将这些独立证据来源聚合在一起,与基线和文献中报道的其他方法相比,会导致预测数量和质量的显著提高。例如,我们的方法为人类生成了超过 12000 个新的蛋白质功能,估计精度约为 76%,其中有 1973 个人类蛋白质的 7500 个新功能注释,这些蛋白质之前只有零个或只有一个功能注释。我们还在一组在结直肠癌中起重要作用的基因(MLH1、PMS2、EPHB4)上验证了我们的预测,根据文献中的证据,我们可以确认其中超过 73%的基因。
将不同的方法组合成一种单一的、综合的预测方法,可以在不同的、但始终保持高精度和非常好的覆盖度的水平上,为分析中包含的每一个物种推断出数千种蛋白质功能。