Helicos BioSciences Corporation, One Kendall Square, Building 700, Cambridge, MA 02139, USA.
BMC Biol. 2010 Dec 21;8:149. doi: 10.1186/1741-7007-8-149.
Discovery that the transcriptional output of the human genome is far more complex than predicted by the current set of protein-coding annotations and that most RNAs produced do not appear to encode proteins has transformed our understanding of genome complexity and suggests new paradigms of genome regulation. However, the fraction of all cellular RNA whose function we do not understand and the fraction of the genome that is utilized to produce that RNA remain controversial. This is not simply a bookkeeping issue because the degree to which this un-annotated transcription is present has important implications with respect to its biologic function and to the general architecture of genome regulation. For example, efforts to elucidate how non-coding RNAs (ncRNAs) regulate genome function will be compromised if that class of RNAs is dismissed as simply 'transcriptional noise'.
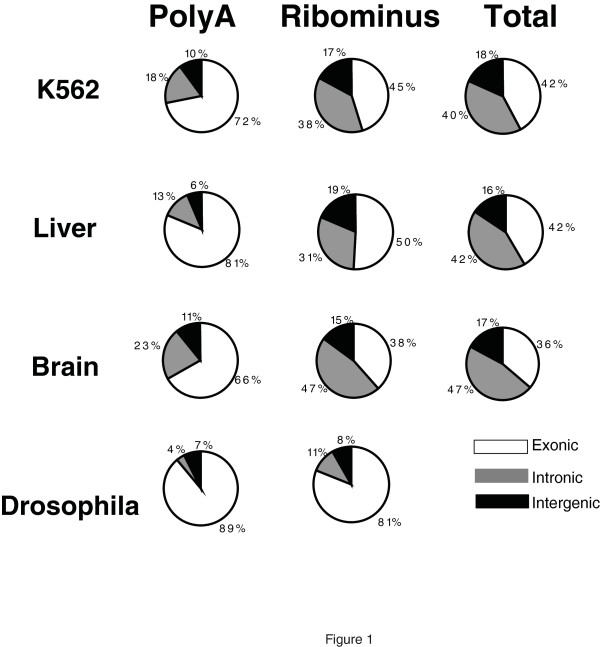
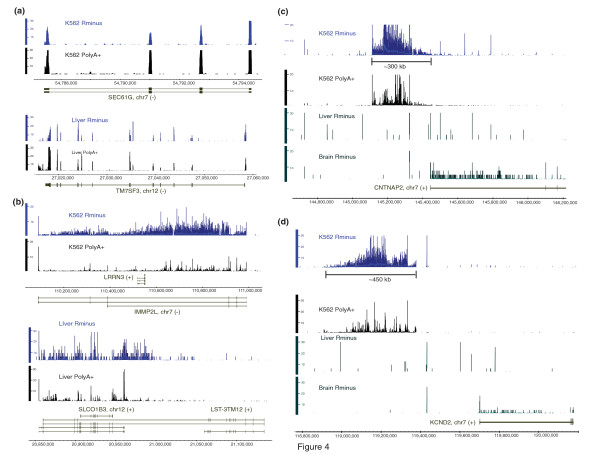
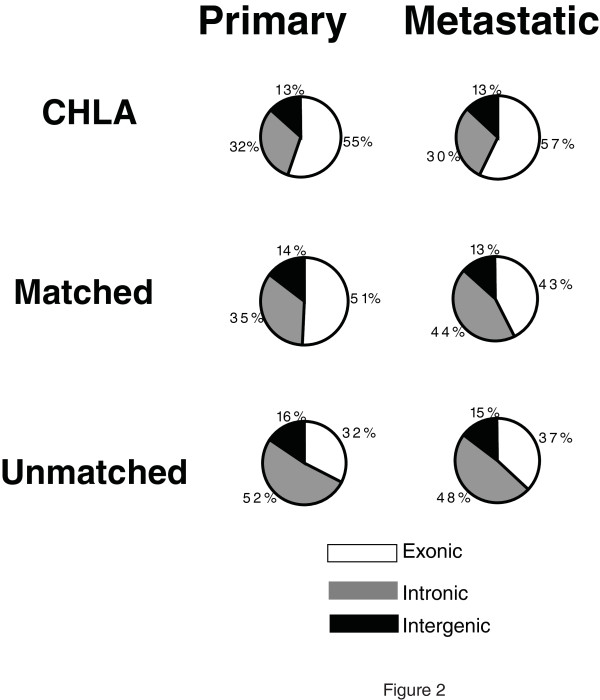
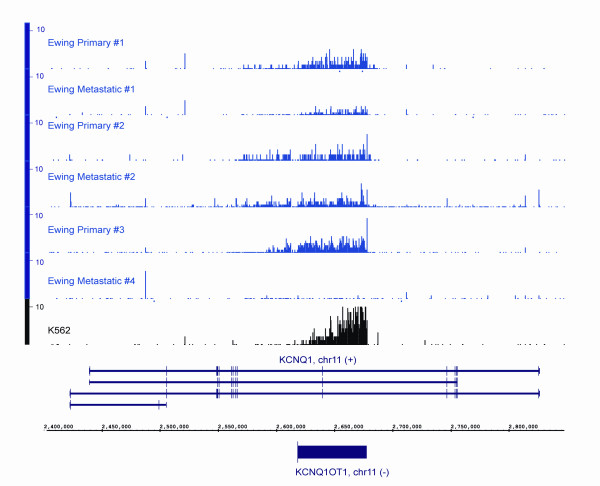
We show that the relative mass of RNA whose function and/or structure we do not understand (the so called 'dark matter' RNAs), as a proportion of all non-ribosomal, non-mitochondrial human RNA (mt-RNA), can be greater than that of protein-encoding transcripts. This observation is obscured in studies that focus only on polyA-selected RNA, a method that enriches for protein coding RNAs and at the same time discards the vast majority of RNA prior to analysis. We further show the presence of a large number of very long, abundantly-transcribed regions (100's of kb) in intergenic space and further show that expression of these regions is associated with neoplastic transformation. These overlap some regions found previously in normal human embryonic tissues and raises an interesting hypothesis as to the function of these ncRNAs in both early development and neoplastic transformation.
We conclude that 'dark matter' RNA can constitute the majority of non-ribosomal, non-mitochondrial-RNA and a significant fraction arises from numerous very long, intergenic transcribed regions that could be involved in neoplastic transformation.
人类基因组的转录输出远比当前蛋白质编码注释集所预测的要复杂,并且大多数产生的 RNA 似乎不编码蛋白质,这一发现改变了我们对基因组复杂性的理解,并提出了新的基因组调控范例。然而,我们不了解其功能的所有细胞 RNA 以及用于产生该 RNA 的基因组的比例仍然存在争议。这不仅仅是一个簿记问题,因为未注释转录的程度对其生物学功能以及基因组调控的一般结构具有重要意义。例如,如果将非编码 RNA(ncRNA)视为简单的“转录噪声”,那么阐明 ncRNA 如何调节基因组功能的努力将受到损害。
我们表明,我们不了解其功能和/或结构的 RNA(所谓的“暗物质”RNA)的相对质量,相对于所有非核糖体、非线粒体人类 RNA(mt-RNA)的比例,可以大于编码蛋白的转录本。如果仅关注聚 A 选择的 RNA 进行研究,这种观察结果就会被掩盖,因为这种方法富集了编码蛋白的 RNA,同时在分析之前丢弃了绝大多数 RNA。我们进一步表明,在基因间空间中存在大量非常长且转录丰富的区域(数百 kb),并且进一步表明这些区域的表达与肿瘤转化有关。这些区域与先前在正常人类胚胎组织中发现的一些区域重叠,并提出了一个有趣的假设,即这些 ncRNA 在早期发育和肿瘤转化中具有功能。
我们得出结论,“暗物质”RNA 可以构成非核糖体、非线粒体 RNA 的大部分,并且很大一部分来自许多非常长的基因间转录区域,这些区域可能与肿瘤转化有关。