ICAR-National Institute for Plant Biotechnology, New Delhi, India.
ICAR-Indian Institute of Pulses Research, Kanpur, Uttar Pradesh, India.
Sci Rep. 2022 Jun 21;12(1):10453. doi: 10.1038/s41598-022-14568-1.
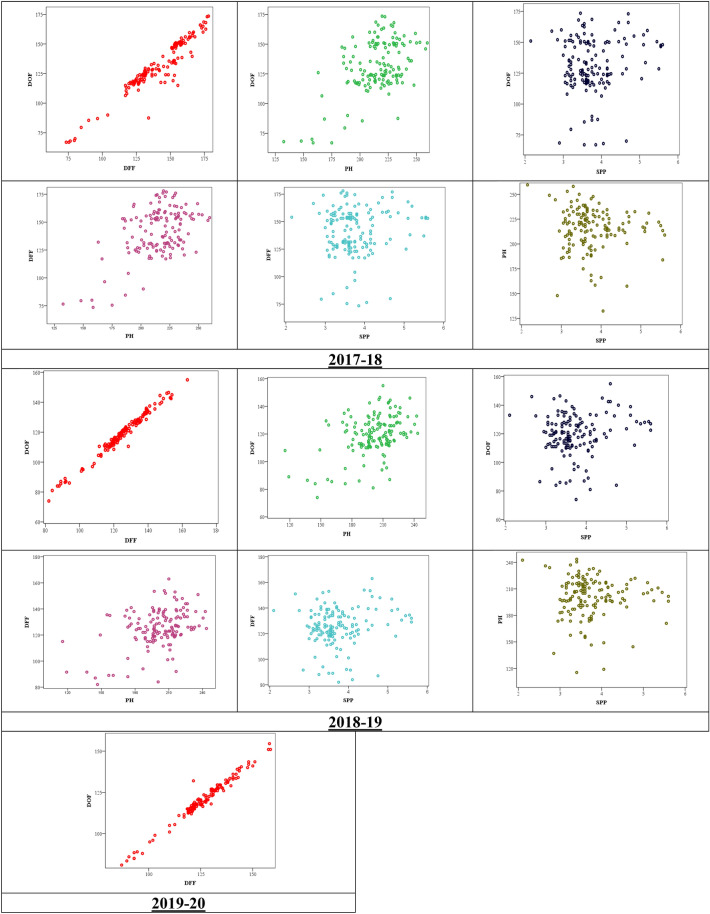
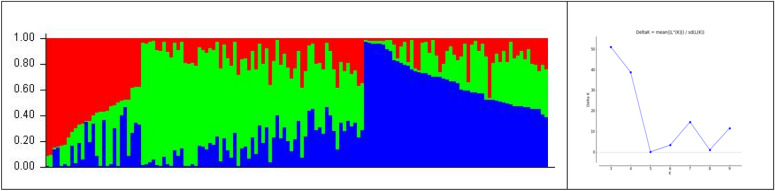
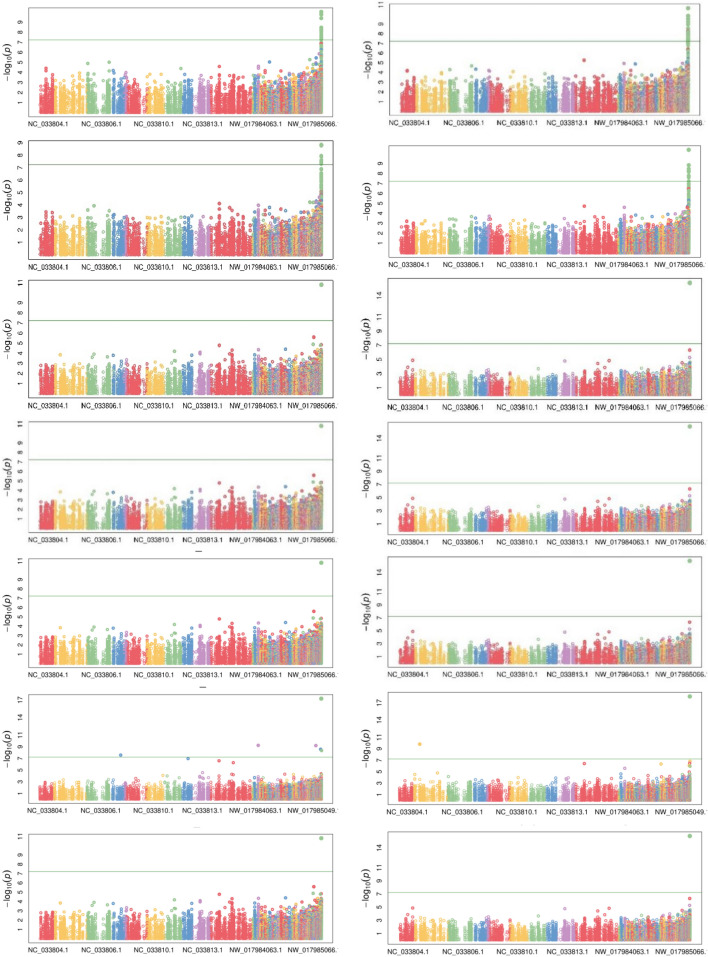
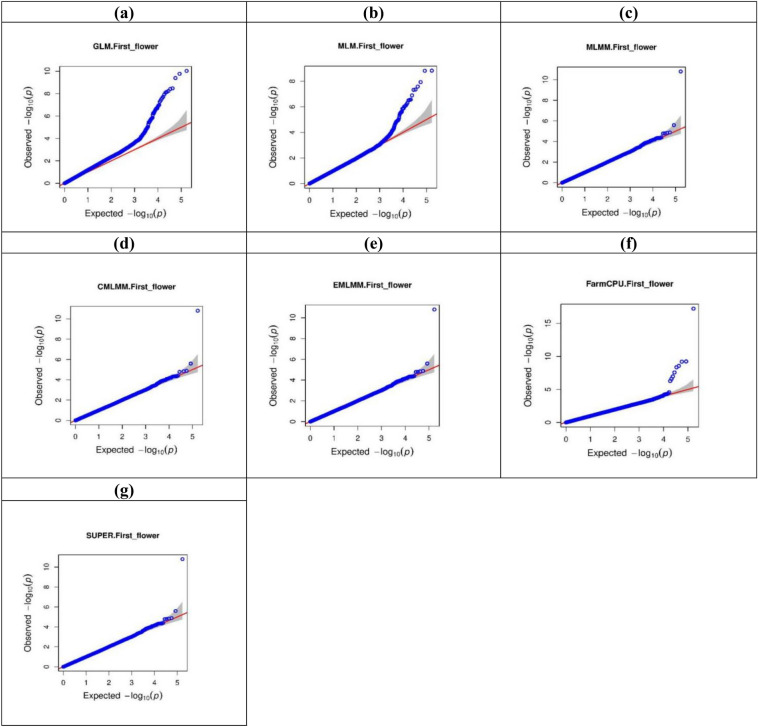
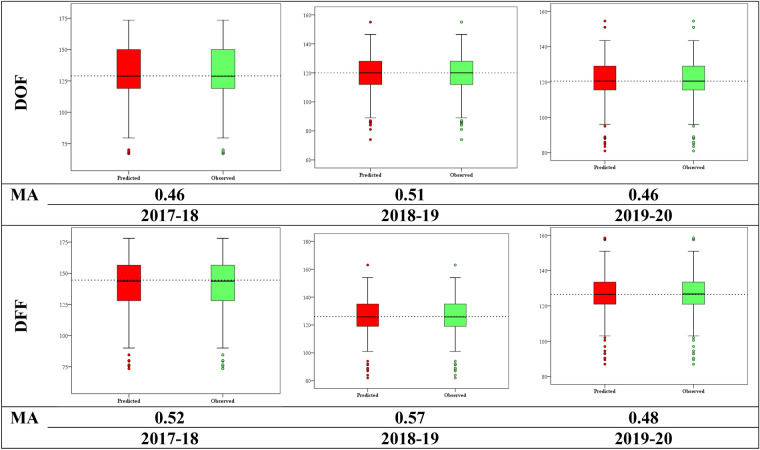
Pigeonpea, a tropical photosensitive crop, harbors significant diversity for days to flowering, but little is known about the genes that govern these differences. Our goal in the current study was to use genome wide association strategy to discover the loci that regulate days to flowering in pigeonpea. A single trait as well as a principal component based association study was conducted on a diverse collection of 142 pigeonpea lines for days to first and fifty percent of flowering over 3 years, besides plant height and number of seeds per pod. The analysis used seven association mapping models (GLM, MLM, MLMM, CMLM, EMLM, FarmCPU and SUPER) and further comparison revealed that FarmCPU is more robust in controlling both false positives and negatives as it incorporates multiple markers as covariates to eliminate confounding between testing marker and kinship. Cumulatively, a set of 22 SNPs were found to be associated with either days to first flowering (DOF), days to fifty percent flowering (DFF) or both, of which 15 were unique to trait based, 4 to PC based GWAS while 3 were shared by both. Because PC1 represents DOF, DFF and plant height (PH), four SNPs found associated to PC1 can be inferred as pleiotropic. A window of ± 2 kb of associated SNPs was aligned with available transcriptome data generated for transition from vegetative to reproductive phase in pigeonpea. Annotation analysis of these regions revealed presence of genes which might be involved in floral induction like Cytochrome p450 like Tata box binding protein, Auxin response factors, Pin like genes, F box protein, U box domain protein, chromatin remodelling complex protein, RNA methyltransferase. In summary, it appears that auxin responsive genes could be involved in regulating DOF and DFF as majority of the associated loci contained genes which are component of auxin signaling pathways in their vicinity. Overall, our findings indicates that the use of principal component analysis in GWAS is statistically more robust in terms of identifying genes and FarmCPU is a better choice compared to the other aforementioned models in dealing with both false positive and negative associations and thus can be used for traits with complex inheritance.
兵豆是一种热带短日照作物,具有丰富的开花时间多样性,但对于控制这些差异的基因知之甚少。我们目前的研究目标是利用全基因组关联策略发现调控兵豆开花时间的基因座。对 142 条兵豆品系进行了为期 3 年的单性状和基于主成分的关联研究,包括开花第一天的天数和开花 50%的天数、株高和每荚种子数。该分析使用了 7 种关联作图模型(GLM、MLM、MLMM、CMLM、EMLM、FarmCPU 和 SUPER),进一步的比较表明,FarmCPU 更稳健,因为它将多个标记作为协变量来控制假阳性和假阴性,从而消除了测试标记和亲缘关系之间的混淆。累积起来,发现了一组 22 个 SNP 与开花第一天的天数(DOF)、开花 50%的天数(DFF)或两者都有关联,其中 15 个是基于性状的,4 个是基于 PC 的 GWAS,3 个是两者共有的。由于 PC1 代表 DOF、DFF 和株高(PH),因此与 PC1 相关的 4 个 SNP 可以推断为多效性。与已发表的兵豆从营养生长到生殖生长转变的转录组数据对齐的相关 SNP 窗口为±2kb。对这些区域的注释分析表明,存在可能参与花诱导的基因,如细胞色素 p450 类塔盒结合蛋白、生长素反应因子、Pin 类基因、F 框蛋白、U 盒结构域蛋白、染色质重塑复合物蛋白、RNA 甲基转移酶。总之,似乎生长素反应基因可能参与调节 DOF 和 DFF,因为大多数相关基因座附近都包含了生长素信号通路的组成基因。总的来说,我们的研究结果表明,在 GWAS 中使用主成分分析在识别基因方面具有统计学上的优势,而且 FarmCPU 是一种比其他上述模型更好的选择,因为它可以处理假阳性和假阴性关联,因此可以用于具有复杂遗传的性状。