The Division of Pulmonary and Critical Care Medicine, Department of Medicine, University of Rochester School of Medicine and Dentistry, Rochester, New York, United States of America.
PLoS One. 2011 Jan 6;6(1):e15909. doi: 10.1371/journal.pone.0015909.
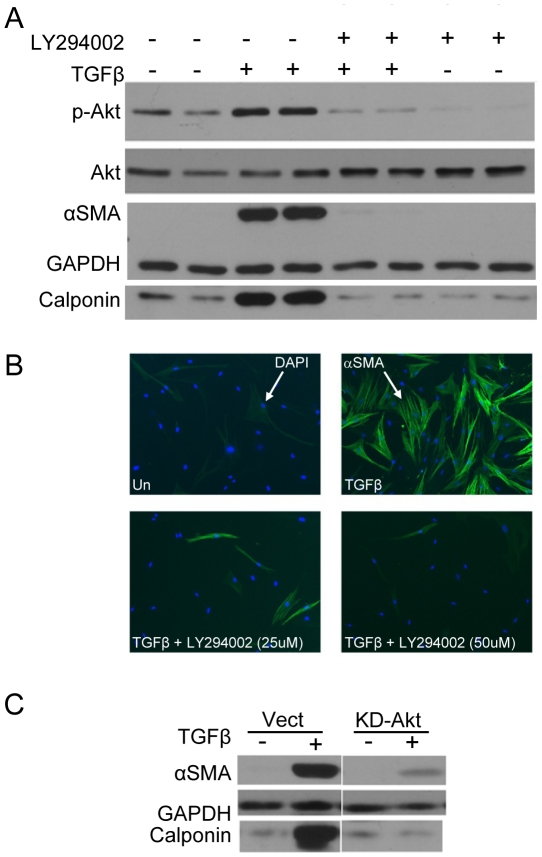
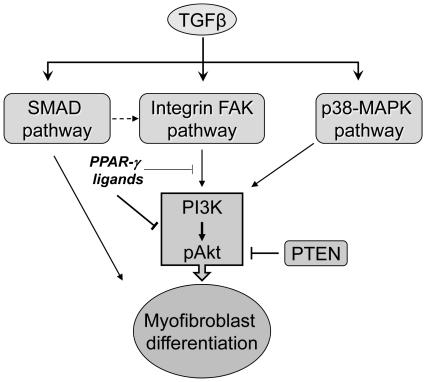
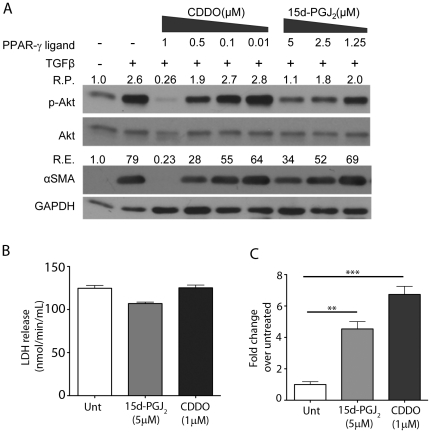
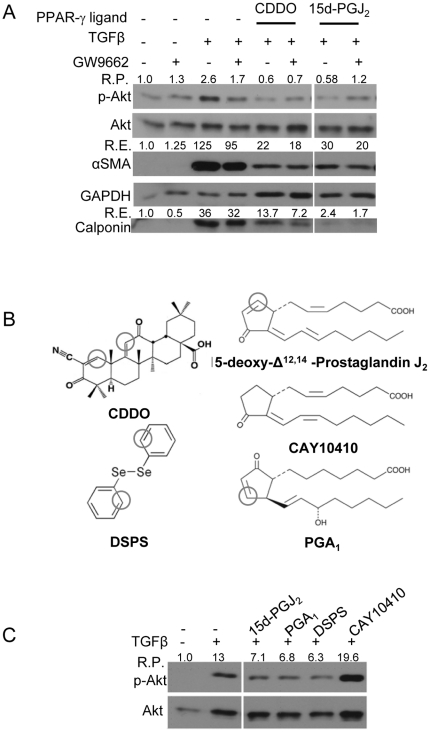
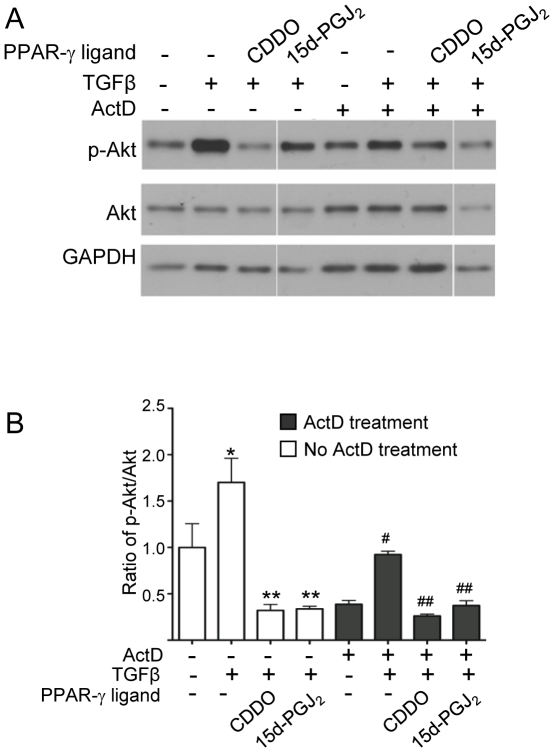
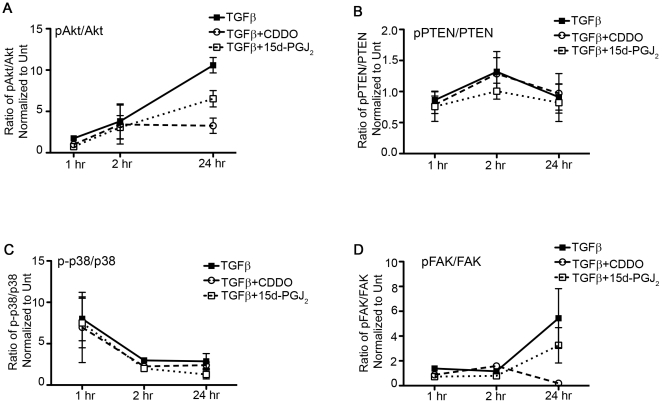
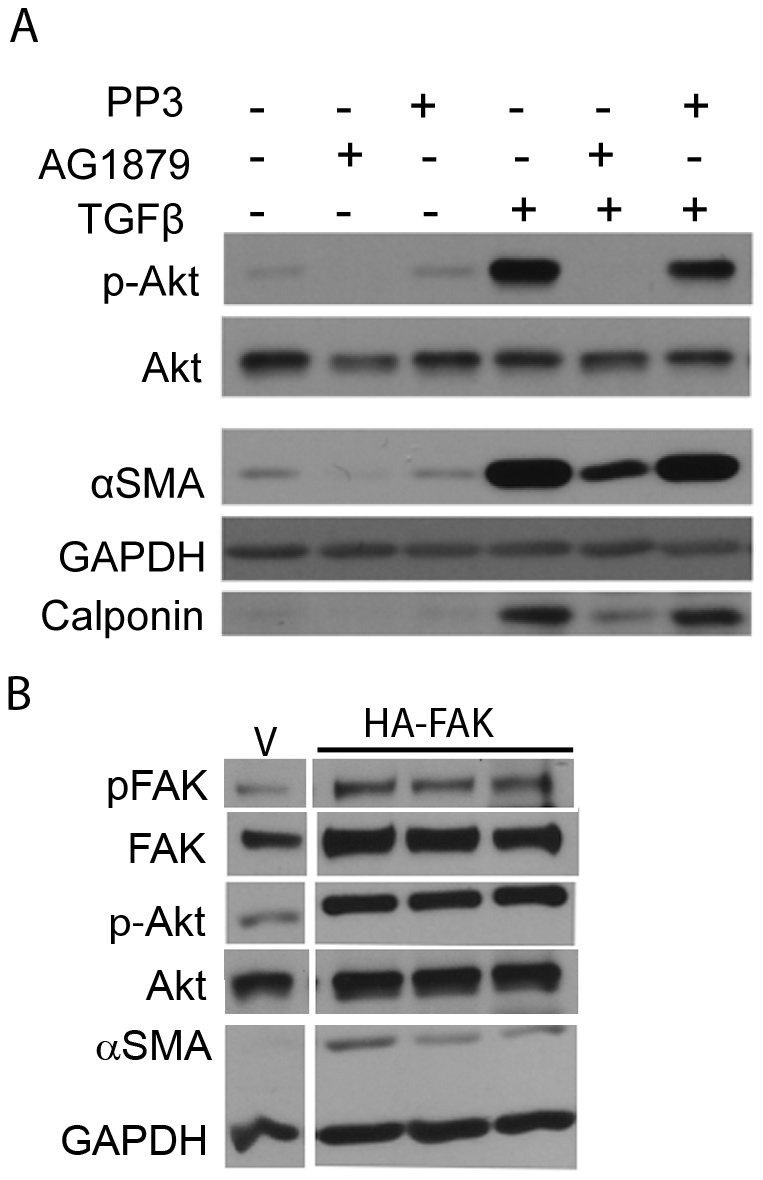
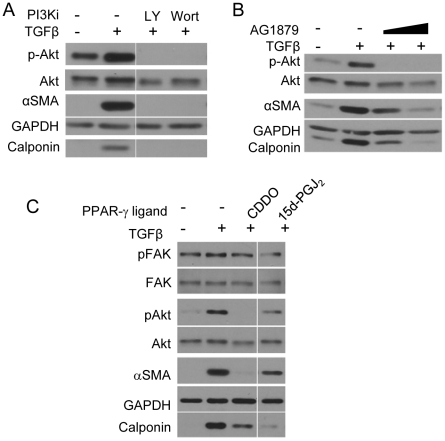
Transforming growth factor beta (TGFβ) induced differentiation of human lung fibroblasts to myofibroblasts is a key event in the pathogenesis of pulmonary fibrosis. Although the typical TGFβ signaling pathway involves the Smad family of transcription factors, we have previously reported that peroxisome proliferator-activated receptor-γ (PPAR-γ) ligands inhibit TGFβ-mediated differentiation of human lung fibroblasts to myofibroblasts via a Smad-independent pathway. TGFβ also activates the phosphatidylinositol 3 kinase/protein kinase B (PI3K/Akt) pathway leading to phosphorylation of Akt(S473). Here, we report that PPAR-γ ligands, 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid (CDDO) and 15-deoxy-(12,14)-15d-prostaglandin J(2) (15d-PGJ(2)), inhibit human myofibroblast differentiation of normal and idiopathic pulmonary fibrotic (IPF) fibroblasts, by blocking Akt phosphorylation at Ser473 by a PPAR-γ-independent mechanism. The PI3K inhibitor LY294002 and a dominant-negative inactive kinase-domain mutant of Akt both inhibited TGFβ-stimulated myofibroblast differentiation, as determined by Western blotting for α-smooth muscle actin and calponin. Prostaglandin A(1) (PGA(1)), a structural analogue of 15d-PGJ(2) with an electrophilic center, also reduced TGFβ-driven phosphorylation of Akt, while CAY10410, another analogue that lacks an electrophilic center, did not; implying that the activity of 15d-PGJ(2) and CDDO is dependent on their electrophilic properties. PPAR-γ ligands inhibited TGFβ-induced Akt phosphorylation via both post-translational and post-transcriptional mechanisms. This inhibition is independent of MAPK-p38 and PTEN but is dependent on TGFβ-induced phosphorylation of FAK, a kinase that acts upstream of Akt. Thus, PPAR-γ ligands inhibit TGFβ signaling by affecting two pro-survival pathways that culminate in myofibroblast differentiation. Further studies of PPAR-γ ligands and small electrophilic molecules may lead to a new generation of anti-fibrotic therapeutics.
转化生长因子-β(TGFβ)诱导人肺成纤维细胞向肌成纤维细胞分化是肺纤维化发病机制中的一个关键事件。虽然典型的 TGFβ信号通路涉及 Smad 家族转录因子,但我们之前的研究报告表明,过氧化物酶体增殖物激活受体-γ(PPAR-γ)配体通过非 Smad 依赖途径抑制 TGFβ介导的人肺成纤维细胞向肌成纤维细胞的分化。TGFβ还激活磷脂酰肌醇 3 激酶/蛋白激酶 B(PI3K/Akt)通路,导致 Akt(S473)的磷酸化。在这里,我们报告 PPAR-γ 配体,2-氰基-3,12-二氧代-1,9-十八烯-28-酸(CDDO)和 15-去氧-(12,14)-15d-前列腺素 J2(15d-PGJ2),通过非 PPAR-γ 独立的机制抑制 Akt 在丝氨酸 473 处的磷酸化,从而抑制正常和特发性肺纤维化(IPF)成纤维细胞的人肌成纤维细胞分化。PI3K 抑制剂 LY294002 和 Akt 的显性失活激酶结构域突变均抑制 TGFβ 刺激的肌成纤维细胞分化,通过 Western blot 检测α-平滑肌肌动蛋白和钙调蛋白确定。前列腺素 A1(PGA1),一种与 15d-PGJ2 具有亲电中心的结构类似物,也降低了 TGFβ 驱动的 Akt 磷酸化,而另一种缺乏亲电中心的类似物 CAY10410 则没有;这意味着 15d-PGJ2 和 CDDO 的活性依赖于它们的亲电性质。PPAR-γ 配体通过翻译后和转录后机制抑制 TGFβ 诱导的 Akt 磷酸化。这种抑制不依赖于 MAPK-p38 和 PTEN,但依赖于 TGFβ 诱导的 FAK 磷酸化,FAK 是一种位于 Akt 上游的激酶。因此,PPAR-γ 配体通过影响两条导致肌成纤维细胞分化的生存途径来抑制 TGFβ 信号。进一步研究 PPAR-γ 配体和小亲电分子可能会导致新一代抗纤维化治疗药物的出现。