Scottish Crop Research Institute, Invergowrie, Dundee, Scotland, UK.
Theor Appl Genet. 2011 May;122(7):1375-83. doi: 10.1007/s00122-011-1538-3. Epub 2011 Jan 26.
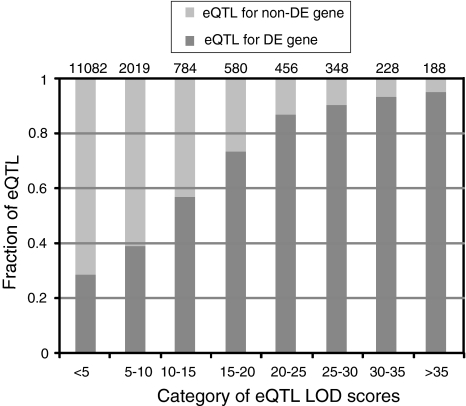
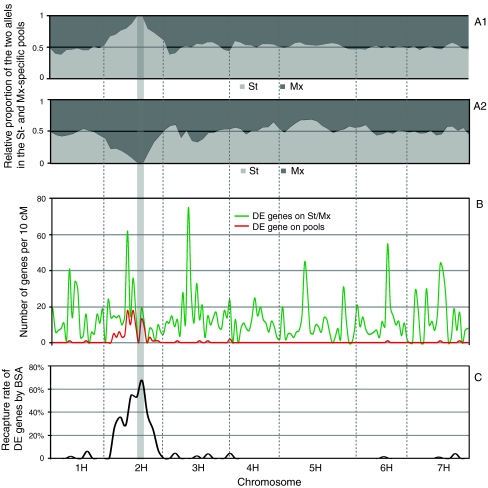
Positional gene isolation in unsequenced species generally requires either a reference genome sequence or an inference of gene content based on conservation of synteny with a genomic model. In the large unsequenced genomes of the Triticeae cereals the latter, i.e. conservation of synteny with the rice and Brachypodium genomes, provides a powerful proxy for establishing local gene content and order. However, efficient exploitation of conservation of synteny requires 'homology bridges' between the model genome and the target region that contains a gene of interest. As effective homology bridges are generally the sequences of genetically mapped genes, increasing the density of these genes around a target locus is an important step in the process. We used bulked segregant analysis (BSA) of transcript abundance data to identify genes located in a specific region of the barley genome. The approach is valuable because only a relatively small proportion of barley genes are currently placed on a genetic map. We analyzed eQTL datasets from the reference Steptoe × Morex doubled haploid population and showed a strong association between differential gene expression and cis-regulation, with 83% of differentially expressed genes co-locating with their eQTL. We then performed BSA by assembling allele-specific pools based on the genotypes of individuals at the partial resistance QTL Rphq11. BSA identified a total of 411 genes as differentially expressed, including HvPHGPx, a gene previously identified as a promising candidate for Rphq11. The genetic location of 276 of these genes could be determined from both eQTL datasets and conservation of synteny, and 254 (92%) of these were located on the target chromosome. We conclude that the identification of differential expression by BSA constitutes a novel method to identify genes located in specific regions of interest. The datasets obtained from such studies provide a robust set of candidate genes for the analysis and serve as valuable resources for targeted marker development and comparative mapping with other grass species.
在未测序的物种中,定位基因通常需要参考基因组序列或基于与基因组模型的共线性来推断基因内容。在大型未测序的禾本科作物基因组中,后者即与水稻和短柄草基因组的共线性,为建立局部基因内容和顺序提供了强大的替代方法。然而,有效地利用共线性需要模型基因组和包含感兴趣基因的目标区域之间的“同源桥”。由于有效的同源桥通常是遗传图谱基因的序列,因此增加目标基因座周围这些基因的密度是该过程的重要步骤。我们使用大量分离群体分析(BSA)的转录丰度数据来鉴定位于大麦基因组特定区域的基因。这种方法很有价值,因为目前只有相对较少的大麦基因被定位在遗传图谱上。我们分析了参考 Steptoe × Morex 加倍单倍体群体的 eQTL 数据集,并显示差异基因表达与顺式调控之间存在很强的关联,83%的差异表达基因与其 eQTL 共定位。然后,我们通过基于部分抗性 QTL Rphq11 个体的基因型组装等位基因特异性池来进行 BSA。BSA 总共鉴定出 411 个差异表达基因,包括 HvPHGPx,这是一个先前被确定为 Rphq11 有前途的候选基因。这些基因中的 276 个的遗传位置可以从 eQTL 数据集和共线性推断中确定,其中 254 个(92%)位于目标染色体上。我们得出结论,BSA 识别差异表达的方法构成了一种鉴定位于特定感兴趣区域的基因的新方法。从这些研究中获得的数据集为分析提供了一组强大的候选基因,并为有针对性的标记开发和与其他草科植物的比较作图提供了有价值的资源。