Gao Feng, Miller J Philip, Xiong Chengjie, Beiser Julia A, Gordon Mae
Division of Biostatistics, Washington University School of Medicine in St. Louis, Campus Box 8067, 660 S. Euclid Ave., St. Louis, MO 63110, USA.
Stat Methods Appt. 2011 Mar 1;20(1):83-100. doi: 10.1007/s10260-010-0150-z.
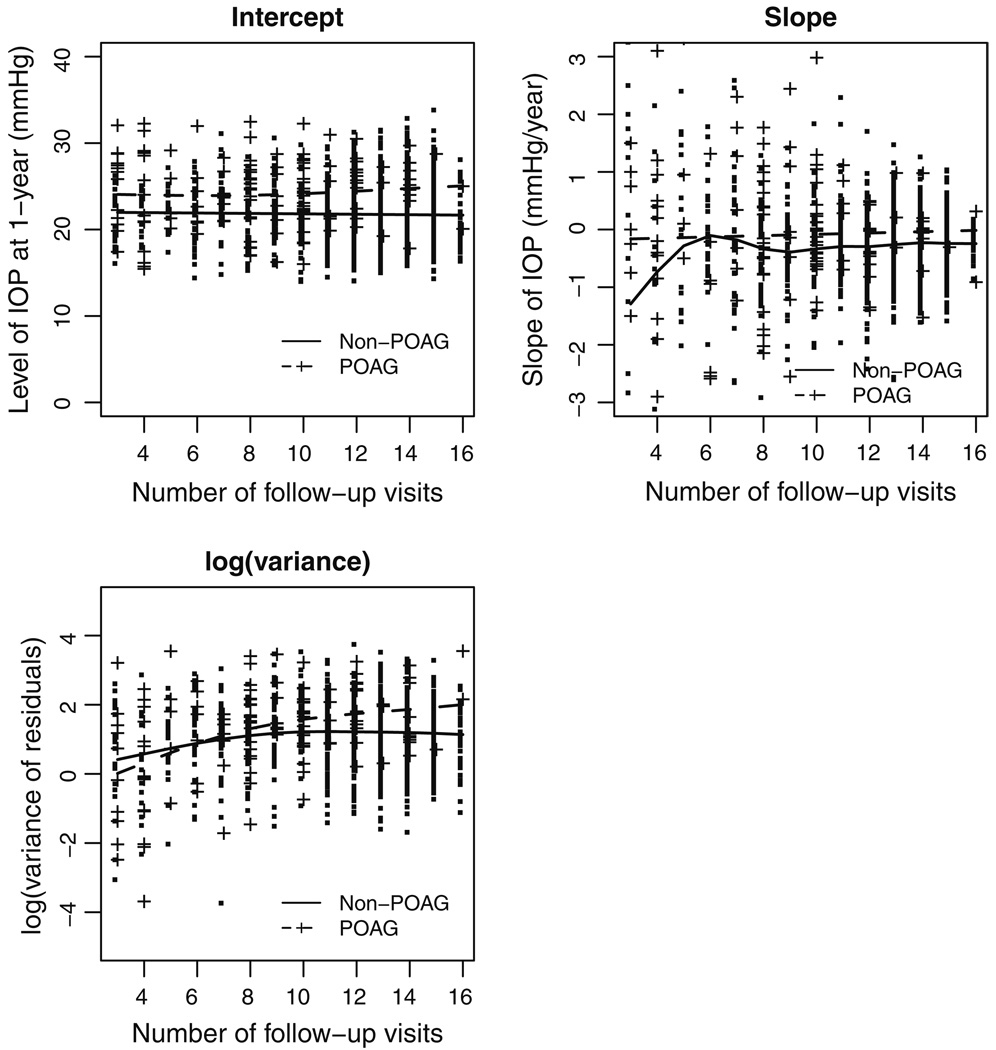
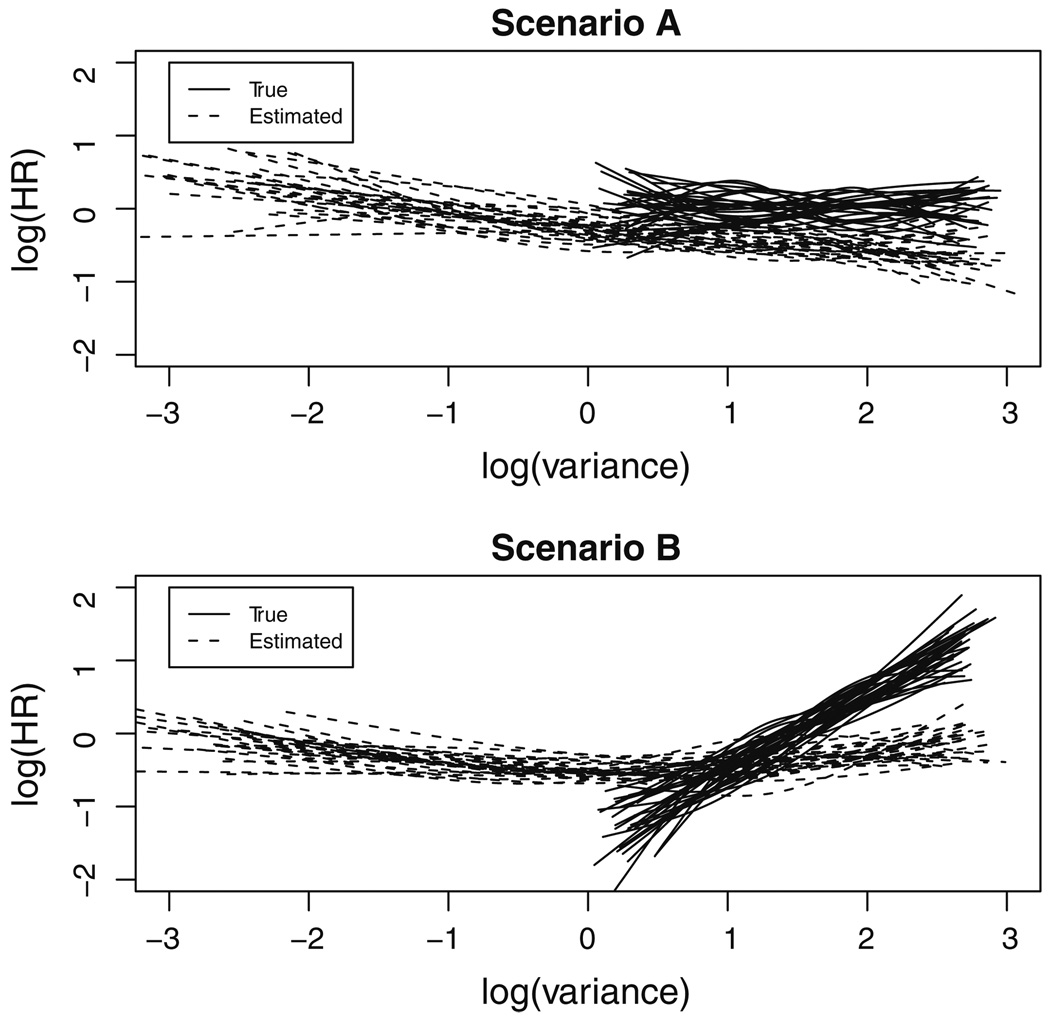
In some clinical trials and epidemiologic studies, investigators are interested in knowing whether the variability of a biomarker is independently predictive of clinical outcomes. This question is often addressed via a naïve approach where a sample-based estimate (e.g., standard deviation) is calculated as a surrogate for the "true" variability and then used in regression models as a covariate assumed to be free of measurement error. However, it is well known that the measurement error in covariates causes underestimation of the true association. The issue of underestimation can be substantial when the precision is low because of limited number of measures per subject. The joint analysis of survival data and longitudinal data enables one to account for the measurement error in longitudinal data and has received substantial attention in recent years. In this paper we propose a joint model to assess the predictive effect of biomarker variability. The joint model consists of two linked sub-models, a linear mixed model with patient-specific variance for longitudinal data and a full parametric Weibull distribution for survival data, and the association between two models is induced by a latent Gaussian process. Parameters in the joint model are estimated under Bayesian framework and implemented using Markov chain Monte Carlo (MCMC) methods with WinBUGS software. The method is illustrated in the Ocular Hypertension Treatment Study to assess whether the variability of intraocular pressure is an independent risk of primary open-angle glaucoma. The performance of the method is also assessed by simulation studies.
在一些临床试验和流行病学研究中,研究者们感兴趣的是了解生物标志物的变异性是否能独立预测临床结局。这个问题通常通过一种简单的方法来解决,即计算基于样本的估计值(如标准差)作为“真实”变异性的替代指标,然后在回归模型中作为假定无测量误差的协变量使用。然而,众所周知,协变量中的测量误差会导致对真实关联的低估。当由于每个受试者的测量次数有限而精度较低时,低估问题可能会很严重。生存数据和纵向数据的联合分析能够考虑纵向数据中的测量误差,并且近年来受到了广泛关注。在本文中,我们提出了一个联合模型来评估生物标志物变异性的预测效果。联合模型由两个相互关联的子模型组成,一个是用于纵向数据的具有患者特异性方差的线性混合模型,另一个是用于生存数据的全参数威布尔分布,两个模型之间的关联由一个潜在高斯过程诱导。联合模型中的参数在贝叶斯框架下进行估计,并使用带有WinBUGS软件的马尔可夫链蒙特卡罗(MCMC)方法来实现。该方法在眼压治疗研究中进行了说明,以评估眼压变异性是否是原发性开角型青光眼的独立风险因素。该方法的性能也通过模拟研究进行了评估。