Department of Biochemistry, University of Zürich, Zürich, Switzerland.
PLoS Comput Biol. 2011 Feb;7(2):e1002002. doi: 10.1371/journal.pcbi.1002002. Epub 2011 Feb 3.
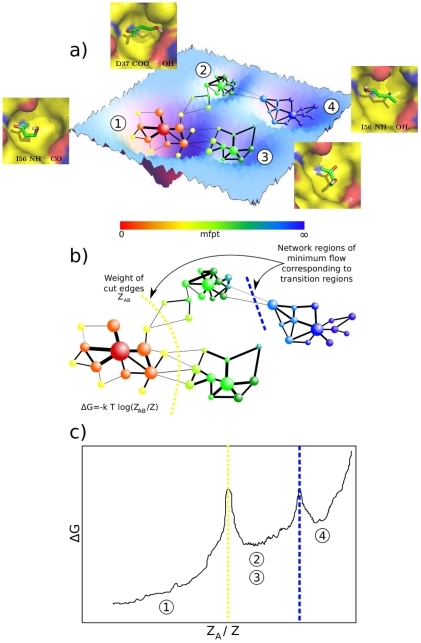
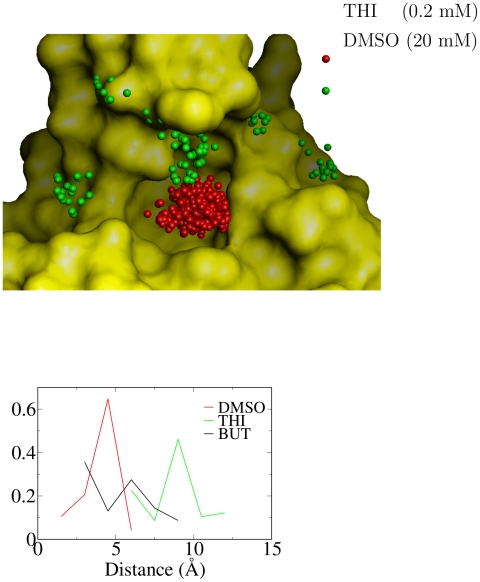
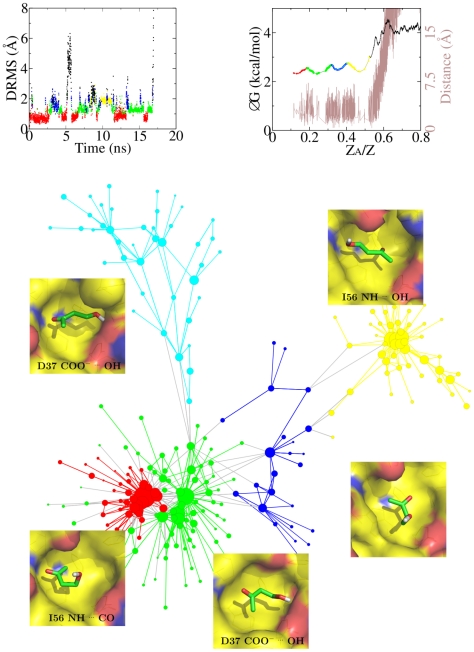
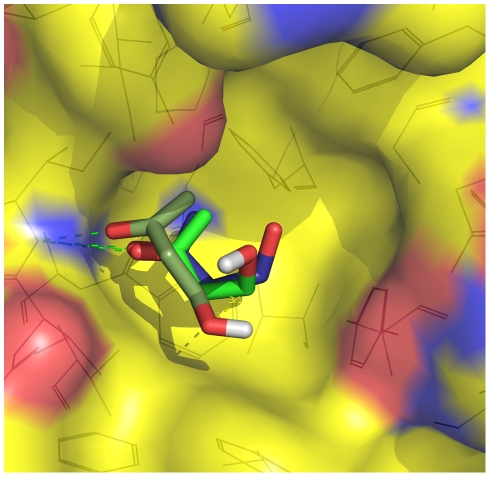
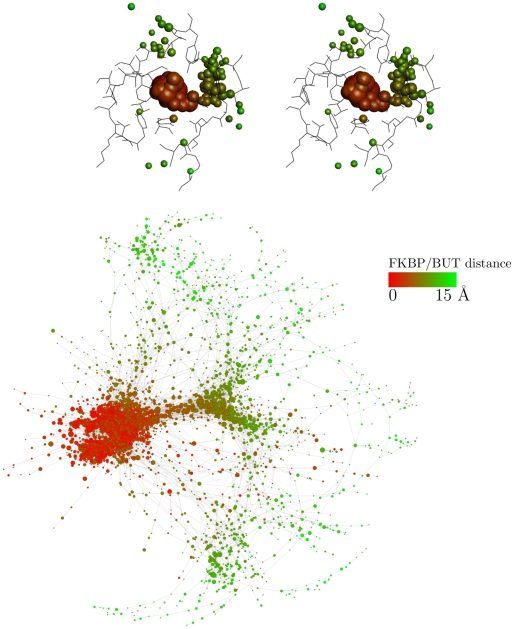
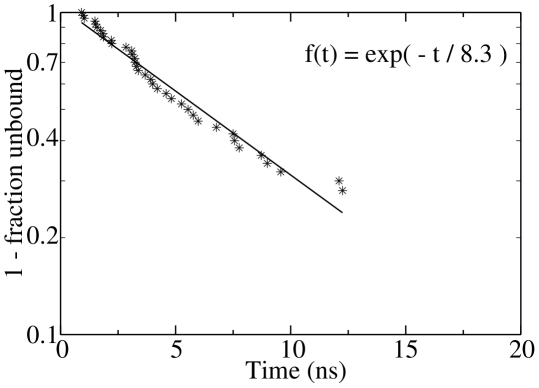
The spontaneous dissociation of six small ligands from the active site of FKBP (the FK506 binding protein) is investigated by explicit water molecular dynamics simulations and network analysis. The ligands have between four (dimethylsulphoxide) and eleven (5-diethylamino-2-pentanone) non-hydrogen atoms, and an affinity for FKBP ranging from 20 to 0.2 mM. The conformations of the FKBP/ligand complex saved along multiple trajectories (50 runs at 310 K for each ligand) are grouped according to a set of intermolecular distances into nodes of a network, and the direct transitions between them are the links. The network analysis reveals that the bound state consists of several subbasins, i.e., binding modes characterized by distinct intermolecular hydrogen bonds and hydrophobic contacts. The dissociation kinetics show a simple (i.e., single-exponential) time dependence because the unbinding barrier is much higher than the barriers between subbasins in the bound state. The unbinding transition state is made up of heterogeneous positions and orientations of the ligand in the FKBP active site, which correspond to multiple pathways of dissociation. For the six small ligands of FKBP, the weaker the binding affinity the closer to the bound state (along the intermolecular distance) are the transition state structures, which is a new manifestation of Hammond behavior. Experimental approaches to the study of fragment binding to proteins have limitations in temporal and spatial resolution. Our network analysis of the unbinding simulations of small inhibitors from an enzyme paints a clear picture of the free energy landscape (both thermodynamics and kinetics) of ligand unbinding.
通过显式水分子动力学模拟和网络分析研究了 FKBP(FK506 结合蛋白)活性部位中六个小分子配体的自发解离。这些配体的非氢原子数介于四个(二甲基亚砜)和十一个(5-二乙氨基-2-戊酮)之间,与 FKBP 的亲和力范围从 20 到 0.2mM。沿着多条轨迹(每个配体 310K 进行 50 次运行)保存的 FKBP/配体复合物的构象根据一组分子间距离被分组到网络的节点中,它们之间的直接跃迁是链接。网络分析表明,结合态由几个亚基组成,即,具有不同分子间氢键和疏水接触的结合模式。解离动力学表现出简单的(即单指数)时间依赖性,因为解键势垒远高于结合态中亚基之间的势垒。解键过渡态由配体在 FKBP 活性部位中的异构位置和取向组成,对应于多种解离途径。对于 FKBP 的六种小分子配体,结合亲和力越弱,过渡态结构越接近结合态(沿着分子间距离),这是 Hammond 行为的新表现。研究片段与蛋白质结合的实验方法在时空分辨率上存在限制。我们对从小型抑制剂从酶中解吸的模拟进行的非键合网络分析清楚地描绘了配体解吸的自由能景观(热力学和动力学)。