Löhrer Rebecca, Neumann-Acikel Aysegül, Eming Rüdiger, Hartmann Karin, Rasokat Heinrich, Krieg Thomas, Happle Rudolf, Eming Sabine
Department of Dermatology, University of Cologne, Cologne, Germany.
Case Rep Dermatol. 2010 Aug 6;2(2):130-4. doi: 10.1159/000319708.
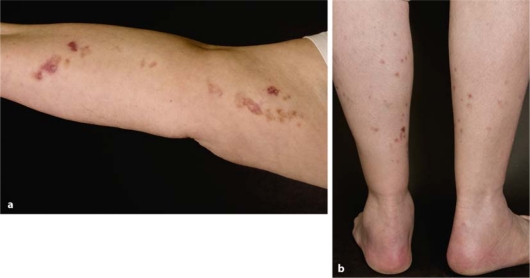
We present a female patient with linear porokeratosis of her right arm since childhood. At the age of 67 years she additionally developed disseminated superficial actinic porokeratosis (DSAP) involving both lower legs. This uncommon coexistence of two different types of porokeratosis fulfils the clinical criteria of a type 2 segmental manifestation of an autosomal dominant skin disorder, being superimposed on the ordinary nonsegmental lesions and reflecting loss of heterozygosity that occurred at an early developmental stage. In DSAP molecular evidence of this concept is so far lacking, but such proof has already been provided in several other autosomal dominant skin disorders. Molecular analysis of cases of type 2 segmental involvement may help elucidate the genetic defect causing DSAP.
我们报告一名女性患者,自童年起右臂患有线状汗孔角化症。67岁时,她双小腿又出现了播散性浅表性光化性汗孔角化症(DSAP)。两种不同类型汗孔角化症的这种罕见共存符合常染色体显性遗传性皮肤病2型节段性表现的临床标准,叠加在普通的非节段性损害上,反映了在发育早期发生的杂合性缺失。在DSAP中,目前尚无这一概念的分子证据,但在其他几种常染色体显性遗传性皮肤病中已经有了此类证据。对2型节段性受累病例的分子分析可能有助于阐明导致DSAP的基因缺陷。