Division of Biostatistics, Department of Clinical Sciences, University of Texas Southwestern Medical Center, Dallas, Texas, United States of America.
PLoS Genet. 2011 Apr;7(4):e1001353. doi: 10.1371/journal.pgen.1001353. Epub 2011 Apr 7.
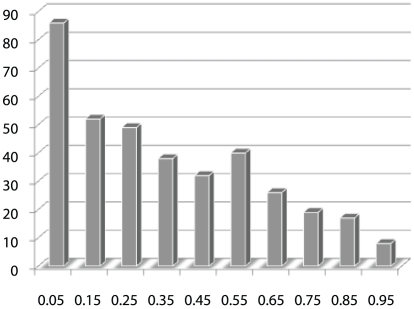
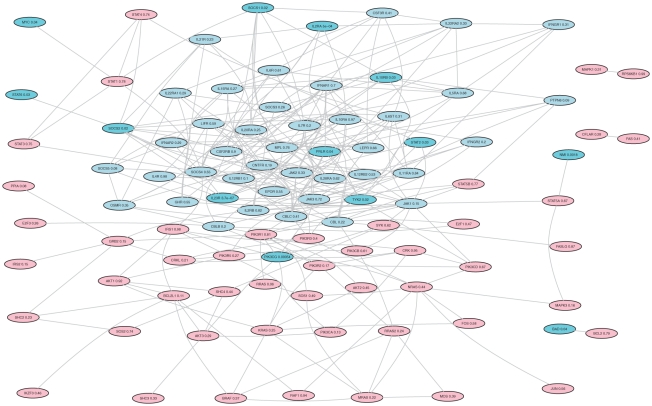
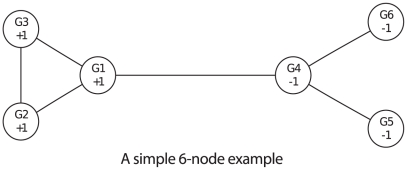
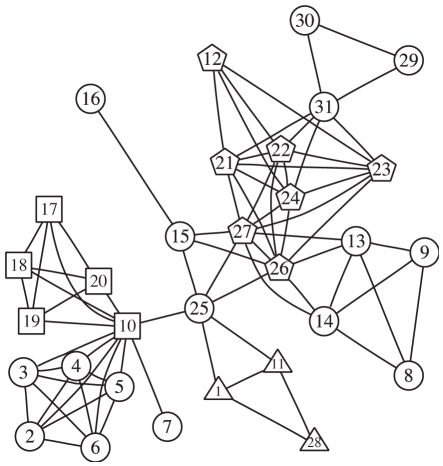
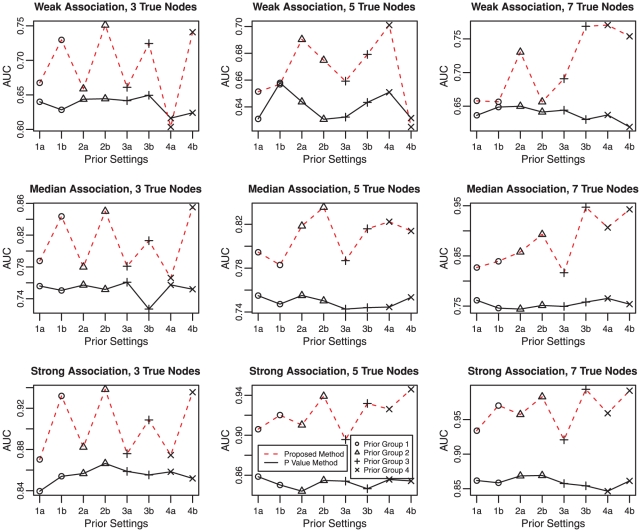
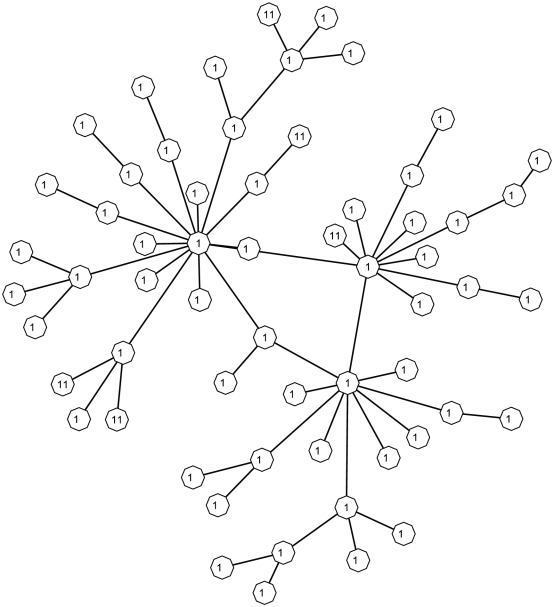
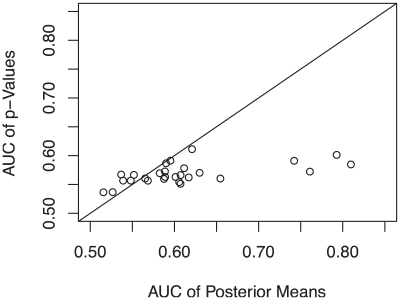
Genome-wide association studies (GWAS) examine a large number of markers across the genome to identify associations between genetic variants and disease. Most published studies examine only single markers, which may be less informative than considering multiple markers and multiple genes jointly because genes may interact with each other to affect disease risk. Much knowledge has been accumulated in the literature on biological pathways and interactions. It is conceivable that appropriate incorporation of such prior knowledge may improve the likelihood of making genuine discoveries. Although a number of methods have been developed recently to prioritize genes using prior biological knowledge, such as pathways, most methods treat genes in a specific pathway as an exchangeable set without considering the topological structure of a pathway. However, how genes are related with each other in a pathway may be very informative to identify association signals. To make use of the connectivity information among genes in a pathway in GWAS analysis, we propose a Markov Random Field (MRF) model to incorporate pathway topology for association analysis. We show that the conditional distribution of our MRF model takes on a simple logistic regression form, and we propose an iterated conditional modes algorithm as well as a decision theoretic approach for statistical inference of each gene's association with disease. Simulation studies show that our proposed framework is more effective to identify genes associated with disease than a single gene-based method. We also illustrate the usefulness of our approach through its applications to a real data example.
全基因组关联研究 (GWAS) 检查基因组中的大量标记,以确定遗传变异与疾病之间的关联。大多数已发表的研究仅检查单一标记,这可能不如同时考虑多个标记和多个基因更具信息量,因为基因可能相互作用以影响疾病风险。文献中积累了大量关于生物途径和相互作用的知识。可以想象,适当纳入这些先验知识可能会提高发现真实结果的可能性。尽管最近已经开发了许多使用先前的生物学知识(如途径)来优先考虑基因的方法,但大多数方法将特定途径中的基因视为可交换的集合,而不考虑途径的拓扑结构。然而,基因之间在途径中的相互关系可能对于识别关联信号非常有帮助。为了在 GWAS 分析中利用途径中基因之间的连接信息,我们提出了一种马尔可夫随机场 (MRF) 模型,以纳入途径拓扑结构进行关联分析。我们表明,我们的 MRF 模型的条件分布采用简单的逻辑回归形式,并且我们提出了一种迭代条件模式算法以及一种决策理论方法,用于统计推断每个基因与疾病的关联。模拟研究表明,与基于单个基因的方法相比,我们提出的框架更有效地识别与疾病相关的基因。我们还通过将其应用于真实数据示例来说明我们方法的有用性。