Department of Computer Science and Center for Bioinformatics and Computational Biology, University of Maryland, College Park, Maryland, USA.
PLoS Comput Biol. 2011 Apr;7(4):e1001119. doi: 10.1371/journal.pcbi.1001119. Epub 2011 Apr 14.
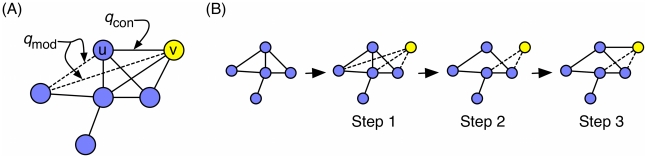
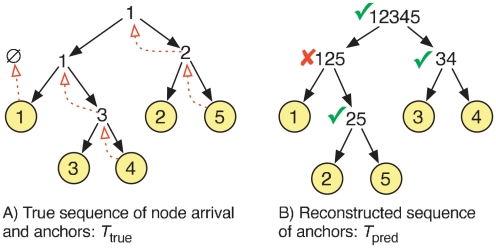
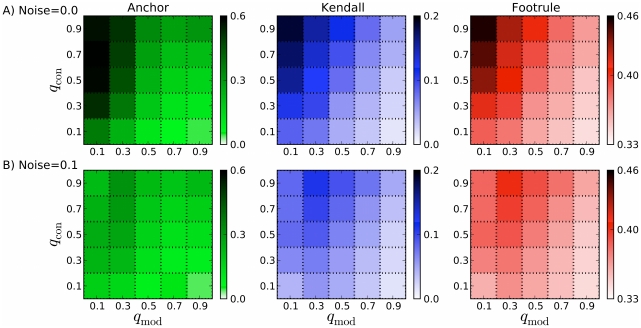
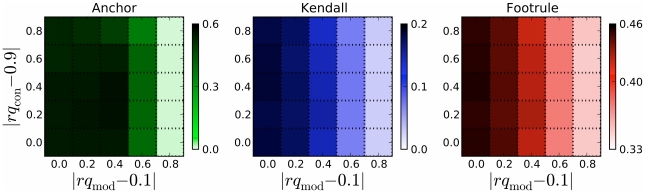
What proteins interacted in a long-extinct ancestor of yeast? How have different members of a protein complex assembled together over time? Our ability to answer such questions has been limited by the unavailability of ancestral protein-protein interaction (PPI) networks. To overcome this limitation, we propose several novel algorithms to reconstruct the growth history of a present-day network. Our likelihood-based method finds a probable previous state of the graph by applying an assumed growth model backwards in time. This approach retains node identities so that the history of individual nodes can be tracked. Using this methodology, we estimate protein ages in the yeast PPI network that are in good agreement with sequence-based estimates of age and with structural features of protein complexes. Further, by comparing the quality of the inferred histories for several different growth models (duplication-mutation with complementarity, forest fire, and preferential attachment), we provide additional evidence that a duplication-based model captures many features of PPI network growth better than models designed to mimic social network growth. From the reconstructed history, we model the arrival time of extant and ancestral interactions and predict that complexes have significantly re-wired over time and that new edges tend to form within existing complexes. We also hypothesize a distribution of per-protein duplication rates, track the change of the network's clustering coefficient, and predict paralogous relationships between extant proteins that are likely to be complementary to the relationships inferred using sequence alone. Finally, we infer plausible parameters for the model, thereby predicting the relative probability of various evolutionary events. The success of these algorithms indicates that parts of the history of the yeast PPI are encoded in its present-day form.
在酵母的一个已灭绝的远古祖先中,有哪些蛋白质相互作用?随着时间的推移,蛋白质复合物的不同成员是如何组装在一起的?由于缺乏祖先蛋白质-蛋白质相互作用(PPI)网络,我们回答这些问题的能力受到了限制。为了克服这一限制,我们提出了几种新的算法来重建当前网络的生长历史。我们的基于似然的方法通过将假设的增长模型反向应用于时间,找到图形的可能先前状态。这种方法保留了节点的身份,以便可以跟踪单个节点的历史。使用这种方法,我们估计了酵母 PPI 网络中蛋白质的年龄,这些年龄与基于序列的年龄估计以及蛋白质复合物的结构特征非常吻合。此外,通过比较几种不同生长模型(互补性复制-突变、森林火灾和优先附着)推断历史的质量,我们提供了额外的证据表明,基于复制的模型比旨在模拟社交网络增长的模型更好地捕捉了 PPI 网络增长的许多特征。从重建的历史中,我们对现存和祖先相互作用的到达时间进行建模,并预测复合物随着时间的推移发生了显著的重新布线,并且新的边缘往往在现有的复合物内形成。我们还假设了每个蛋白质的复制率分布,跟踪网络聚类系数的变化,并预测现存蛋白质之间的旁系关系,这些关系很可能与仅使用序列推断的关系互补。最后,我们推断模型的合理参数,从而预测各种进化事件的相对概率。这些算法的成功表明,酵母 PPI 的部分历史信息被编码在其当前形式中。