Institute of Plant Science and Resources, Okayama University, Kurashiki, 710-0046, Japan.
BMC Genomics. 2011 May 19;12:246. doi: 10.1186/1471-2164-12-246.
Genome sequencing of barley has been delayed due to its large genome size (ca. 5,000 Mbp). Among the fast sequencing systems, 454 liquid phase pyrosequencing provides the longest reads and is the most promising method for BAC clones. Here we report the results of pooled sequencing of BAC clones selected with ESTs genetically mapped to chromosome 3H.
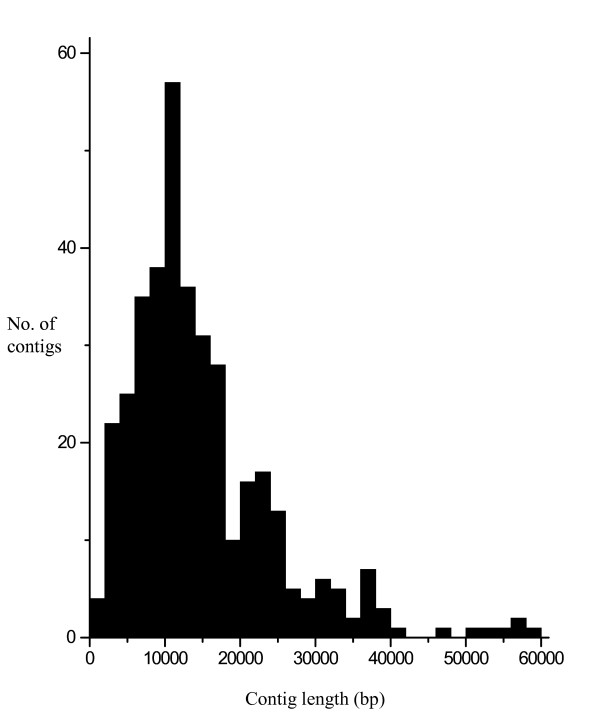
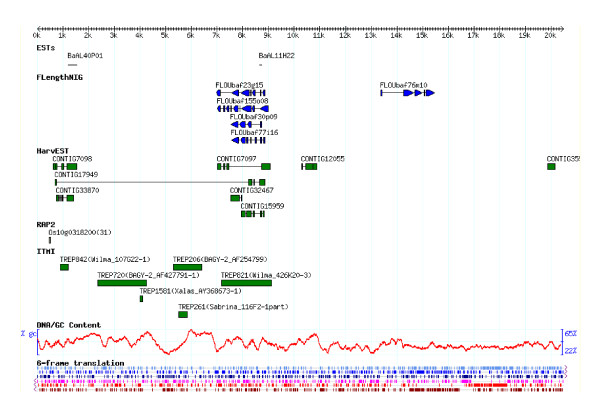
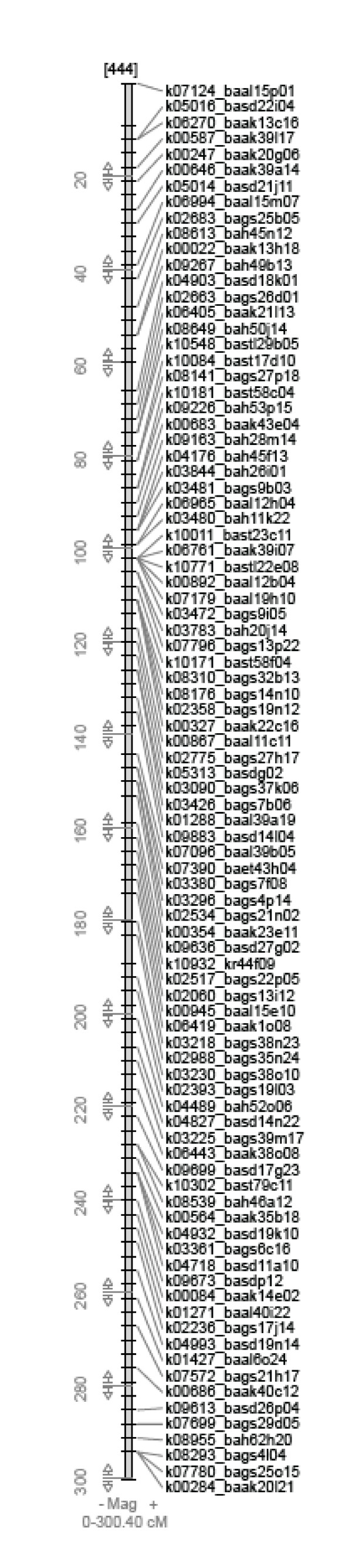
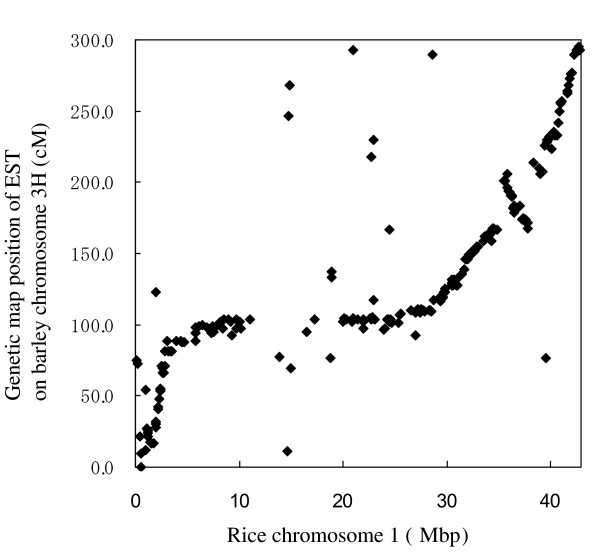
We sequenced pooled barley BAC clones using a 454 parallel genome sequencer. A PCR screening system based on primer sets derived from genetically mapped ESTs on chromosome 3H was used for clone selection in a BAC library developed from cultivar "Haruna Nijo". The DNA samples of 10 or 20 BAC clones were pooled and used for shotgun library development. The homology between contig sequences generated in each pooled library and mapped EST sequences was studied. The number of contigs assigned on chromosome 3H was 372. Their lengths ranged from 1,230 bp to 58,322 bp with an average 14,891 bp. Of these contigs, 240 showed homology and colinearity with the genome sequence of rice chromosome 1. A contig annotation browser supplemented with query search by unique sequence or genetic map position was developed. The identified contigs can be annotated with barley cDNAs and reference sequences on the browser. Homology analysis of these contigs with rice genes indicated that 1,239 rice genes can be assigned to barley contigs by the simple comparison of sequence lengths in both species. Of these genes, 492 are assigned to rice chromosome 1.
We demonstrate the efficiency of sequencing gene rich regions from barley chromosome 3H, with special reference to syntenic relationships with rice chromosome 1.
由于大麦基因组大小(约 5000 Mbp)较大,其基因组测序一直被延迟。在快速测序系统中,454 液相焦磷酸测序提供了最长的读长,是 BAC 克隆的最有前途的方法。在这里,我们报告了使用 EST 遗传图谱定位到 3H 染色体的 BAC 克隆进行池测序的结果。
我们使用 454 平行基因组测序仪对 pooled barley BAC 克隆进行测序。基于来自“Haruna Nijo”品种染色体 3H 上遗传图谱的 EST 衍生的引物组,我们建立了一个 PCR 筛选系统,用于从构建的 BAC 文库中选择克隆。将 10 或 20 个 BAC 克隆的 DNA 样品混合用于 shotgun 文库的开发。研究了每个 pooled 文库中生成的 contig 序列与映射 EST 序列之间的同源性。在每个 pooled 文库中分配到染色体 3H 的 contig 数为 372。它们的长度从 1230 bp 到 58322 bp 不等,平均为 14891 bp。在这些 contig 中,有 240 个与水稻 1 号染色体的基因组序列具有同源性和共线性。开发了一个带有查询搜索的 contig 注释浏览器,该浏览器可以通过唯一序列或遗传图谱位置进行查询搜索。在浏览器中,可以对鉴定的 contig 进行大麦 cDNA 和参考序列的注释。这些 contig 与水稻基因的同源性分析表明,通过比较两种物种的序列长度,可以将 1239 个水稻基因分配到大麦 contig 中。其中,有 492 个基因被分配到水稻 1 号染色体。
我们展示了从大麦 3H 染色体测序富含基因的区域的效率,特别参考了与水稻 1 号染色体的同线性关系。