Plant Breeding Department, Wageningen University and Research Centre, Wageningen, The Netherlands.
BMC Bioinformatics. 2011 May 19;12:172. doi: 10.1186/1471-2105-12-172.
Automated genotype calling in tetraploid species was until recently not possible, which hampered genetic analysis. Modern genotyping assays often produce two signals, one for each allele of a bi-allelic marker. While ample software is available to obtain genotypes (homozygous for either allele, or heterozygous) for diploid species from these signals, such software is not available for tetraploid species which may be scored as five alternative genotypes (aaaa, baaa, bbaa, bbba and bbbb; nulliplex to quadruplex).
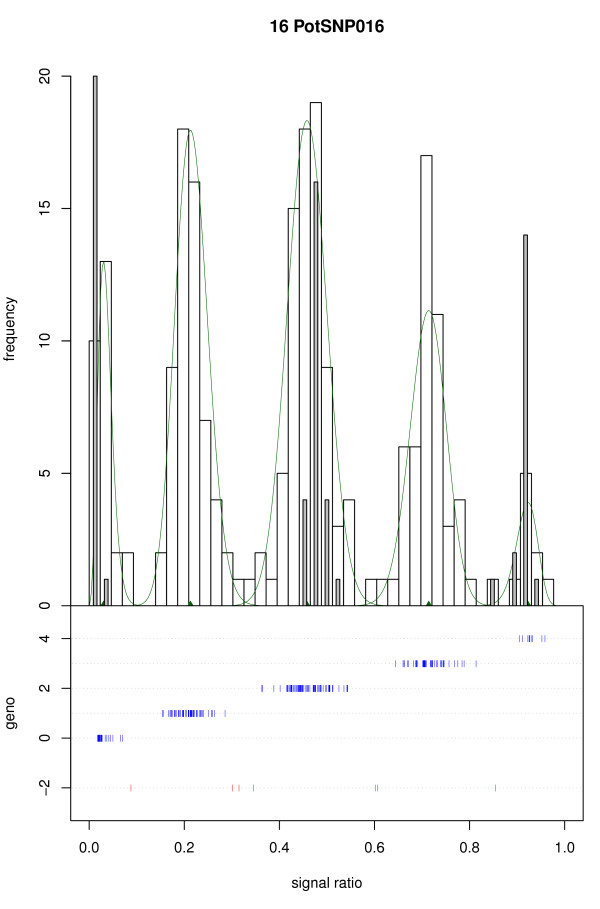
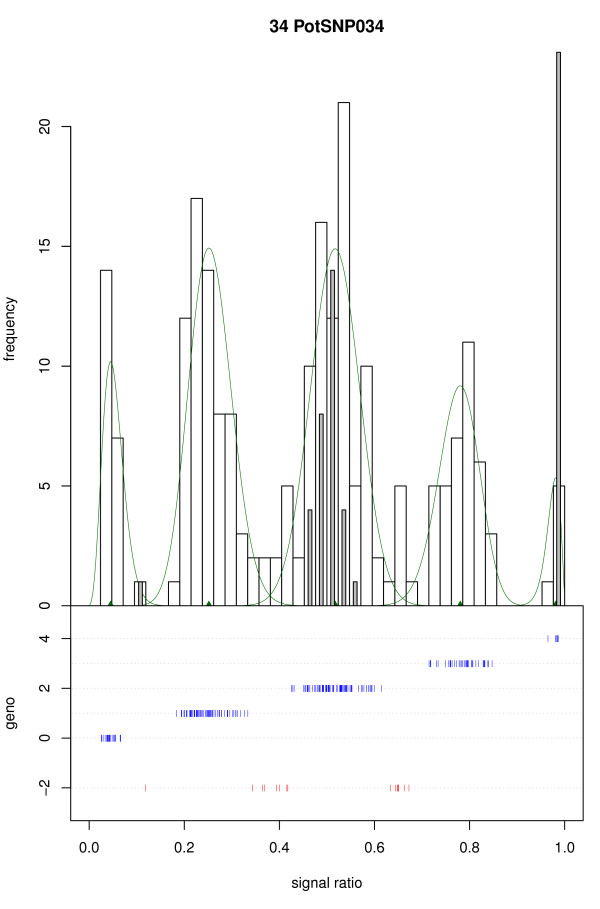
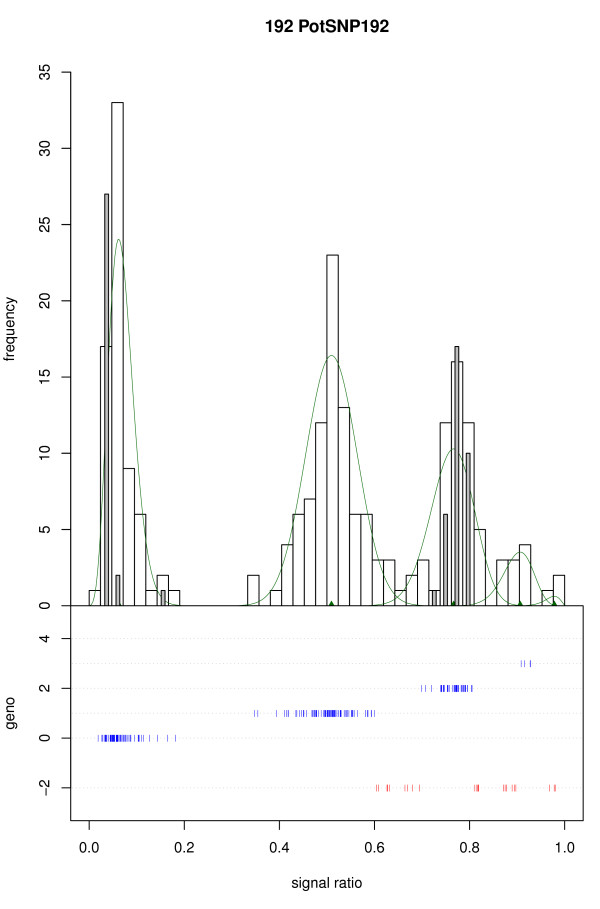
We present a novel algorithm, implemented in the R package fitTetra, to assign genotypes for bi-allelic markers to tetraploid samples from genotyping assays that produce intensity signals for both alleles. The algorithm is based on the fitting of several mixture models with five components, one for each of the five possible genotypes. The models have different numbers of parameters specifying the relation between the five component means, and some of them impose a constraint on the mixing proportions to conform to Hardy-Weinberg equilibrium (HWE) ratios. The software rejects markers that do not allow a reliable genotyping for the majority of the samples, and it assigns a missing score to samples that cannot be scored into one of the five possible genotypes with sufficient confidence.
We have validated the software with data of a collection of 224 potato varieties assayed with an Illumina GoldenGate™ 384 SNP array and shown that all SNPs with informative ratio distributions are fitted. Almost all fitted models appear to be correct based on visual inspection and comparison with diploid samples. When the collection of potato varieties is analyzed as if it were a population, almost all markers seem to be in Hardy-Weinberg equilibrium. The R package fitTetra is freely available under the GNU Public License from http://www.plantbreeding.wur.nl/UK/software_fitTetra.html and as Additional files with this article.
直到最近,四倍体物种的自动化基因型调用都不可能,这阻碍了遗传分析。现代基因分型检测通常会产生两个信号,每个等位基因一个。虽然有大量的软件可用于从这些信号中获得二倍体物种的基因型(纯合子或杂合子),但对于可能被评为五种替代基因型(aaaa、baaa、bbaa、bbba 和 bbbb;单倍体到四倍体)的四倍体物种,没有这样的软件。
我们提出了一种新算法,该算法在 R 包 fitTetra 中实现,用于从产生两个等位基因强度信号的基因分型检测中为二倍体标记分配基因型到四倍体样本。该算法基于拟合五个成分的几个混合模型,每个成分对应于五种可能基因型中的一种。模型具有不同数量的参数,用于指定五个成分均值之间的关系,其中一些模型对混合比例施加约束,以符合 Hardy-Weinberg 平衡(HWE)比例。该软件拒绝大多数样本无法进行可靠基因分型的标记,并对无法以足够置信度将样本分为五种可能基因型之一的样本分配缺失分数。
我们使用 224 个马铃薯品种的 Illumina GoldenGate™ 384 SNP 阵列数据进行了软件验证,并表明所有具有信息比率分布的 SNP 都可以拟合。几乎所有拟合的模型似乎都是正确的,这基于目视检查和与二倍体样本的比较。当马铃薯品种集合被分析为一个群体时,几乎所有标记似乎都处于 Hardy-Weinberg 平衡状态。R 包 fitTetra 可根据 GNU 公共许可证免费获得,网址为 http://www.plantbreeding.wur.nl/UK/software_fitTetra.html,并随本文的其他文件提供。