Chair of Bioinformatics, Boku University Vienna, Muthgasse 18, 1190 Vienna, Austria.
BMC Bioinformatics. 2011 May 19;12:173. doi: 10.1186/1471-2105-12-173.
Sequence analysis aims to identify biologically relevant signals against a backdrop of functionally meaningless variation. Increasingly, it is recognized that the quality of the background model directly affects the performance of analyses. State-of-the-art approaches rely on classical sequence models that are adapted to the studied dataset. Although performing well in the analysis of globular protein domains, these models break down in regions of stronger compositional bias or low complexity. While these regions are typically filtered, there is increasing anecdotal evidence of functional roles. This motivates an exploration of more complex sequence models and application-specific approaches for the investigation of biased regions.
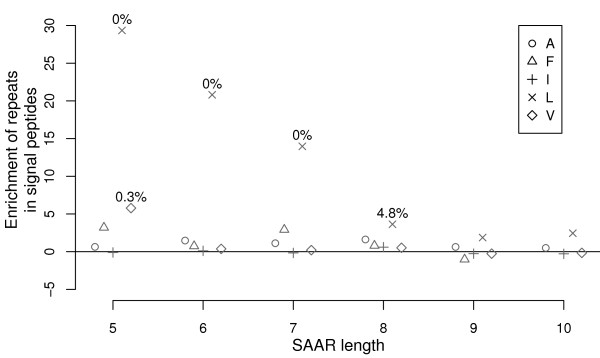
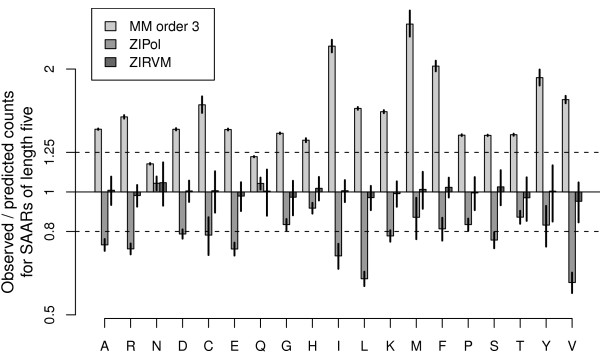
Traditional Markov-chains and application-specific regression models are compared using the example of predicting runs of single amino acids, a particularly simple class of biased regions. Cross-fold validation experiments reveal that the alternative regression models capture the multi-variate trends well, despite their low dimensionality and in contrast even to higher-order Markov-predictors. We show how the significance of unusual observations can be computed for such empirical models. The power of a dedicated model in the detection of biologically interesting signals is then demonstrated in an analysis identifying the unexpected enrichment of contiguous leucine-repeats in signal-peptides. Considering different reference sets, we show how the question examined actually defines what constitutes the 'background'. Results can thus be highly sensitive to the choice of appropriate model training sets. Conversely, the choice of reference data determines the questions that can be investigated in an analysis.
Using a specific case of studying biased regions as an example, we have demonstrated that the construction of application-specific background models is both necessary and feasible in a challenging sequence analysis situation.
序列分析旨在识别生物学相关信号,同时排除功能上无意义的变异。越来越多的人认识到,背景模型的质量直接影响分析的性能。最先进的方法依赖于经典的序列模型,这些模型适用于所研究的数据集。虽然在球状蛋白结构域的分析中表现良好,但这些模型在组成性偏差较大或复杂度较低的区域失效。虽然这些区域通常会被过滤掉,但越来越多的轶事证据表明它们具有功能作用。这促使我们探索更复杂的序列模型和特定于应用的方法,以研究有偏差的区域。
使用预测单个氨基酸连续出现的例子,比较了传统的马尔可夫链和特定于应用的回归模型,这是一类特别简单的有偏差区域。交叉验证实验表明,尽管替代回归模型的维度较低,甚至与高阶马尔可夫预测器相比,它们仍能很好地捕捉多变量趋势。我们展示了如何为这种经验模型计算异常观测的显著性。然后,在一个分析中,我们展示了一个专门的模型在检测生物有趣信号方面的强大功能,该分析确定了信号肽中连续亮氨酸重复的意外富集。考虑到不同的参考集,我们展示了所研究的问题实际上如何定义“背景”。因此,结果对适当模型训练集的选择高度敏感。相反,参考数据的选择决定了在分析中可以研究的问题。
以研究有偏差区域的具体案例为例,我们已经证明,在具有挑战性的序列分析情况下,构建特定于应用的背景模型不仅是必要的,而且是可行的。