MOE Key Laboratory of Contemporary Anthropology, School of Life Sciences, Fudan University, Shanghai, 200433, China.
BMC Genomics. 2011 Jun 17;12:322. doi: 10.1186/1471-2164-12-322.
Next-generation sequencing technologies can effectively detect the entire spectrum of genomic variation and provide a powerful tool for systematic exploration of the universe of common, low frequency and rare variants in the entire genome. However, the current paradigm for genome-wide association studies (GWAS) is to catalogue and genotype common variants (5% < MAF). The methods and study design for testing the association of low frequency (0.5% < MAF ≤ 5%) and rare variation (MAF ≤ 0.5%) have not been thoroughly investigated. The 1000 Genomes Project represents one such endeavour to characterize the human genetic variation pattern at the MAF = 1% level as a foundation for association studies. In this report, we explore different strategies and study designs for the near future GWAS in the post-era, based on both low coverage pilot data and exon pilot data in 1000 Genomes Project.
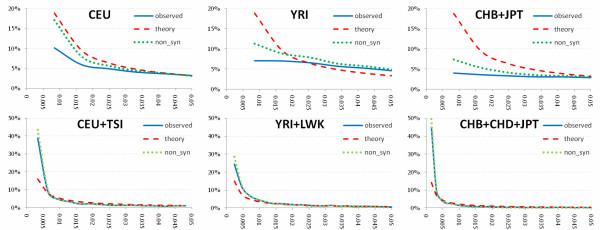
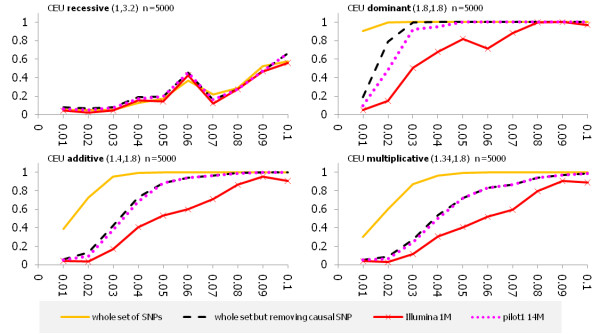
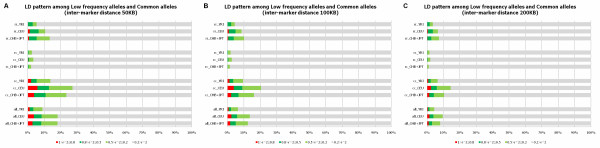
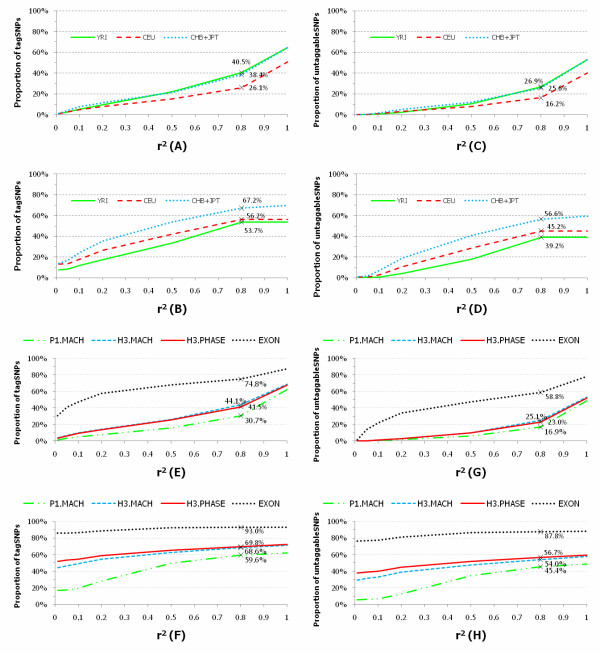
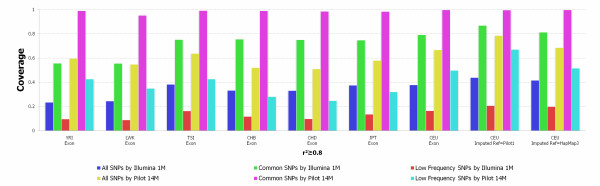
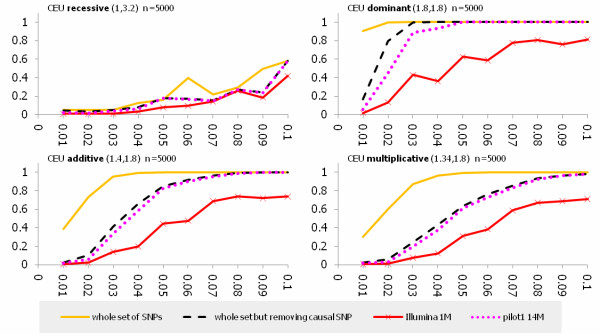
We investigated the linkage disequilibrium (LD) pattern among common and low frequency SNPs and its implication for association studies. We found that the LD between low frequency alleles and low frequency alleles, and low frequency alleles and common alleles are much weaker than the LD between common and common alleles. We examined various tagging designs with and without statistical imputation approaches and compare their power against de novo resequencing in mapping causal variants under various disease models. We used the low coverage pilot data which contain ~14 M SNPs as a hypothetical genotype-array platform (Pilot 14 M) to interrogate its impact on the selection of tag SNPs, mapping coverage and power of association tests. We found that even after imputation we still observed 45.4% of low frequency SNPs which were untaggable and only 67.7% of the low frequency variation was covered by the Pilot 14 M array.
This suggested GWAS based on SNP arrays would be ill-suited for association studies of low frequency variation.
下一代测序技术可以有效地检测基因组变异的全貌,为系统探索全基因组常见、低频和稀有变异提供了强大的工具。然而,目前全基因组关联研究(GWAS)的范例是对常见变异(MAF<5%)进行编目和基因分型。低频(0.5%<MAF≤5%)和稀有变异(MAF≤0.5%)关联检验的方法和研究设计尚未得到彻底研究。1000 基因组计划(The 1000 Genomes Project)就是这样一个旨在以 MAF=1%的水平描述人类遗传变异模式的项目,作为关联研究的基础。在本报告中,我们基于 1000 基因组计划中的低覆盖率试点数据和外显子试点数据,探索了后 GWAS 时代的不同策略和研究设计。
我们研究了常见和低频 SNPs 之间的连锁不平衡(LD)模式及其对关联研究的影响。我们发现,低频等位基因与低频等位基因之间,以及低频等位基因与常见等位基因之间的 LD 比常见等位基因与常见等位基因之间的 LD 弱得多。我们检查了各种带有和不带有统计推断方法的标记设计,并比较了它们在各种疾病模型下对因果变异进行映射的能力与从头重测序。我们使用包含约 1400 万个 SNPs 的低覆盖率试点数据作为假设的基因型阵列平台(Pilot 14 M),研究其对标记 SNP 选择、映射覆盖和关联检验功效的影响。我们发现,即使在推断之后,我们仍然观察到 45.4%的低频 SNP 是无法标记的,而且 Pilot 14 M 阵列只覆盖了 67.7%的低频变异。
这表明基于 SNP 阵列的 GWAS 不适合低频变异的关联研究。