MRC Centre for Outbreak Analysis and Modelling, Department of Infectious Disease Epidemiology, Imperial College London, Faculty of Medicine, London, United Kingdom.
PLoS Comput Biol. 2013;9(10):e1003254. doi: 10.1371/journal.pcbi.1003254. Epub 2013 Oct 10.
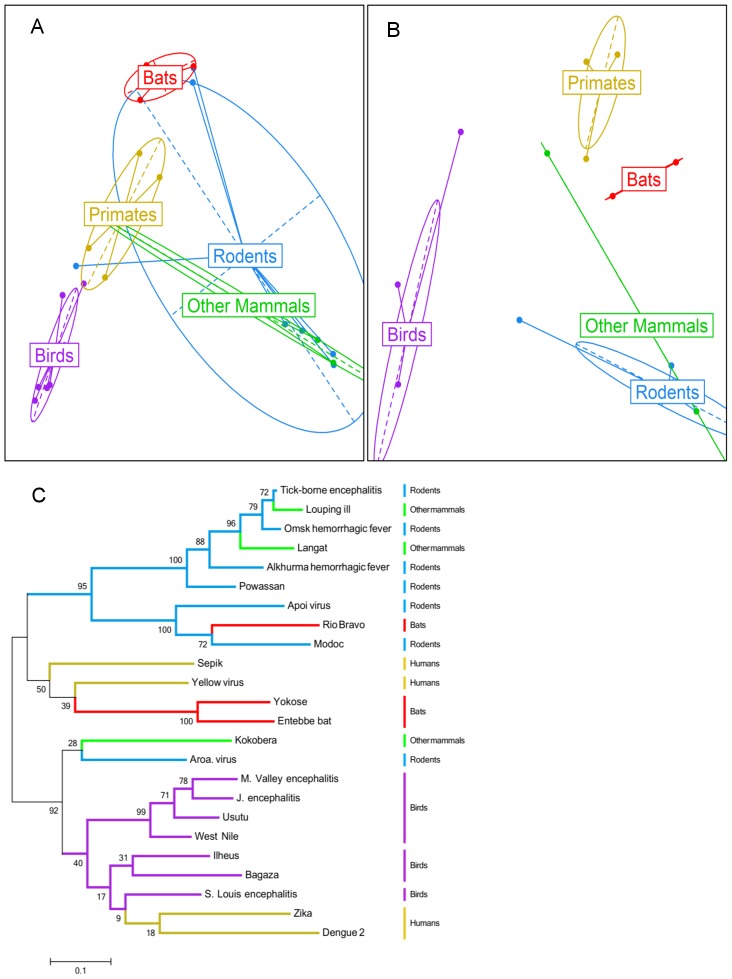
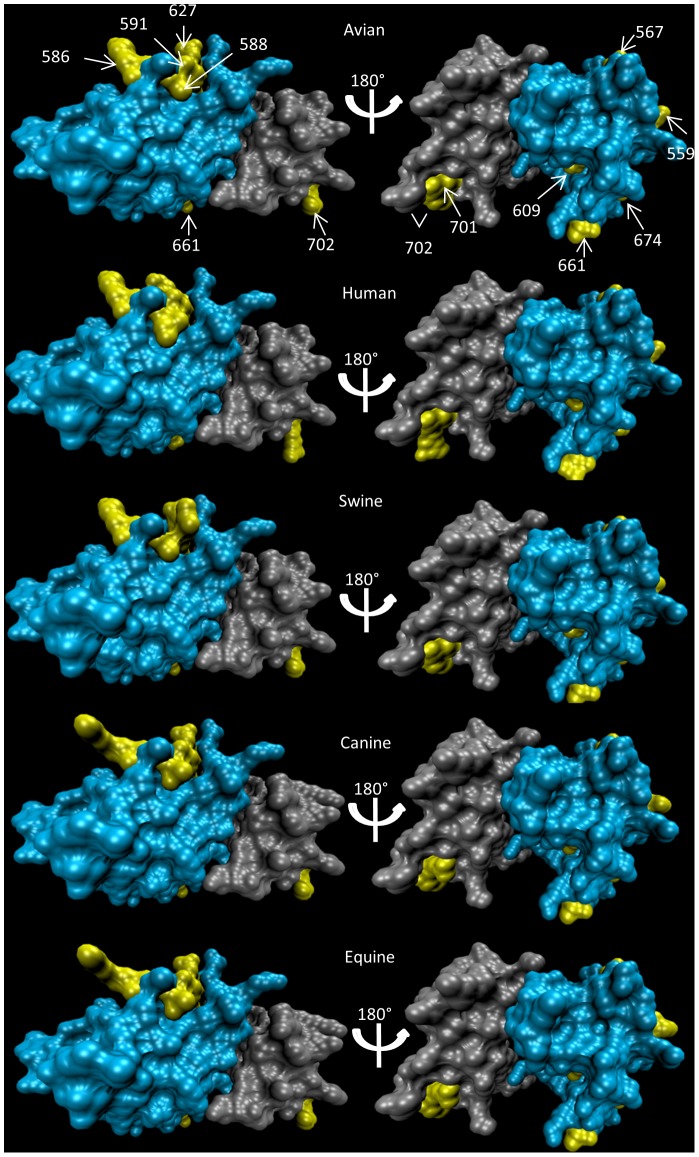
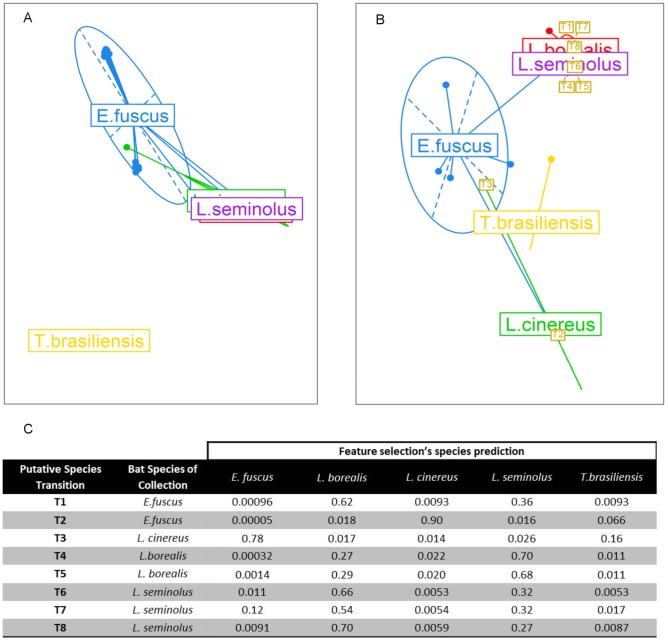
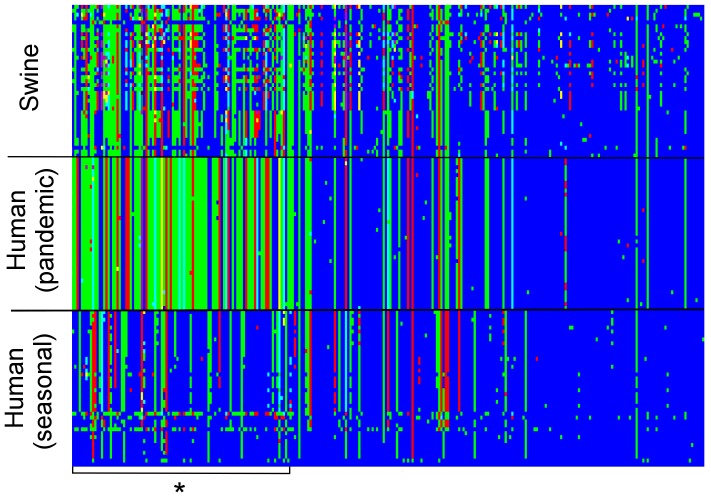
Despite environmental, social and ecological dependencies, emergence of zoonotic viruses in human populations is clearly also affected by genetic factors which determine cross-species transmission potential. RNA viruses pose an interesting case study given their mutation rates are orders of magnitude higher than any other pathogen--as reflected by the recent emergence of SARS and Influenza for example. Here, we show how feature selection techniques can be used to reliably classify viral sequences by host species, and to identify the crucial minority of host-specific sites in pathogen genomic data. The variability in alleles at those sites can be translated into prediction probabilities that a particular pathogen isolate is adapted to a given host. We illustrate the power of these methods by: 1) identifying the sites explaining SARS coronavirus differences between human, bat and palm civet samples; 2) showing how cross species jumps of rabies virus among bat populations can be readily identified; and 3) de novo identification of likely functional influenza host discriminant markers.
尽管存在环境、社会和生态方面的依存关系,但人畜共患病毒在人类群体中的出现显然也受到决定跨物种传播潜力的遗传因素的影响。鉴于 RNA 病毒的突变率比任何其他病原体都要高几个数量级——例如最近 SARS 和流感的出现,它们是一个有趣的案例研究。在这里,我们展示了如何使用特征选择技术可靠地按宿主物种对病毒序列进行分类,并确定病原体基因组数据中关键的少数宿主特异性位点。这些位点的等位基因的可变性可以转化为预测特定病原体分离株是否适应特定宿主的概率。我们通过以下方式说明了这些方法的强大功能:1)鉴定解释 SARS 冠状病毒在人类、蝙蝠和棕榈狸样本之间差异的位点;2)展示如何轻松识别狂犬病病毒在蝙蝠种群之间的跨物种跳跃;3)从头鉴定可能的流感宿主判别标记。