Nelson Sarah C, Doheny Kimberly F, Pugh Elizabeth W, Romm Jane M, Ling Hua, Laurie Cecelia A, Browning Sharon R, Weir Bruce S, Laurie Cathy C
Department of Biostatistics, University of Washington, Seattle, Washington, 98195.
G3 (Bethesda). 2013 Oct 3;3(10):1795-807. doi: 10.1534/g3.113.007161.
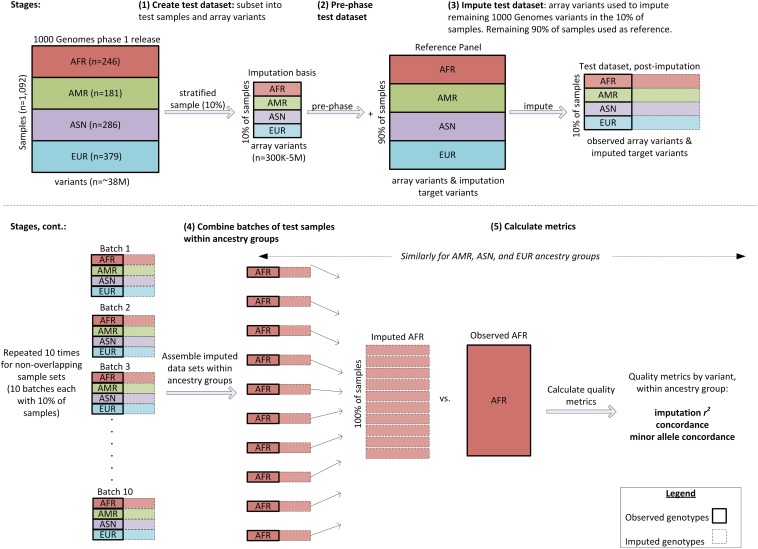
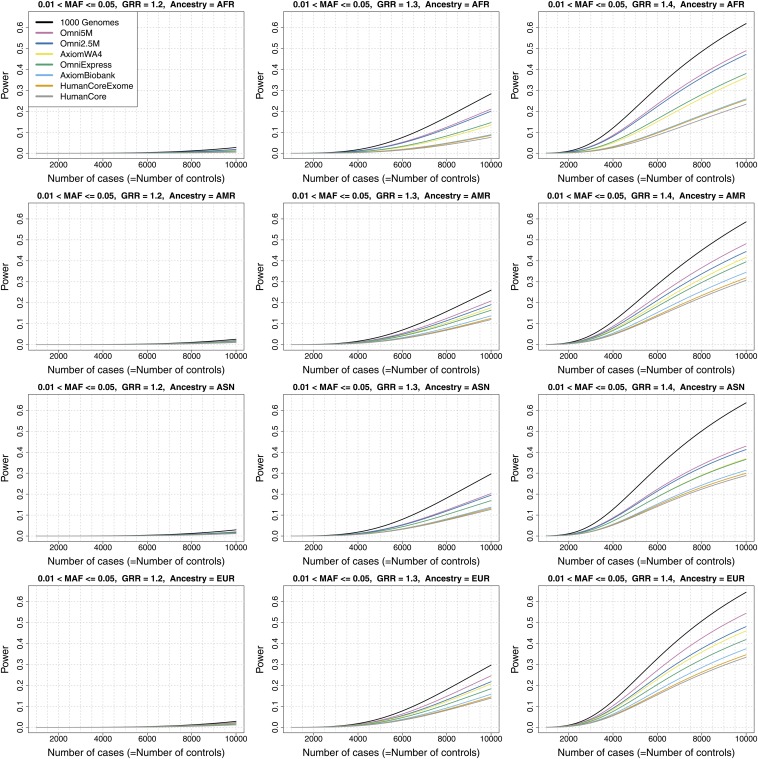
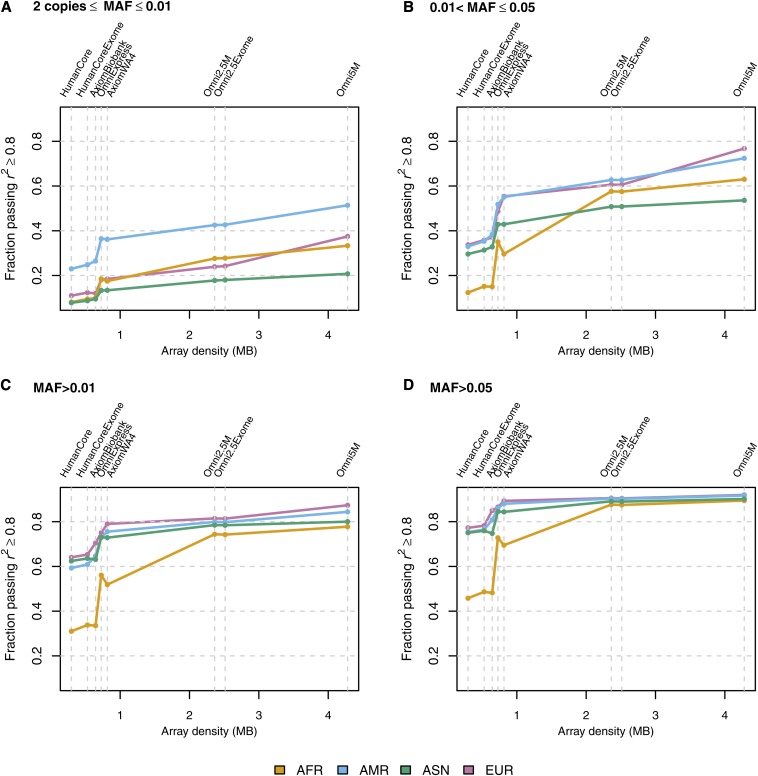
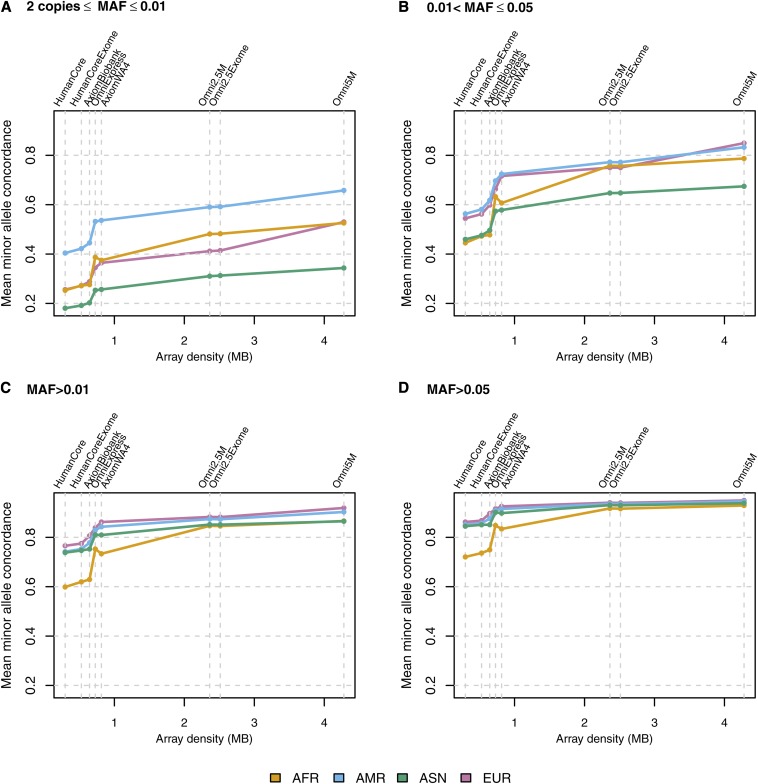
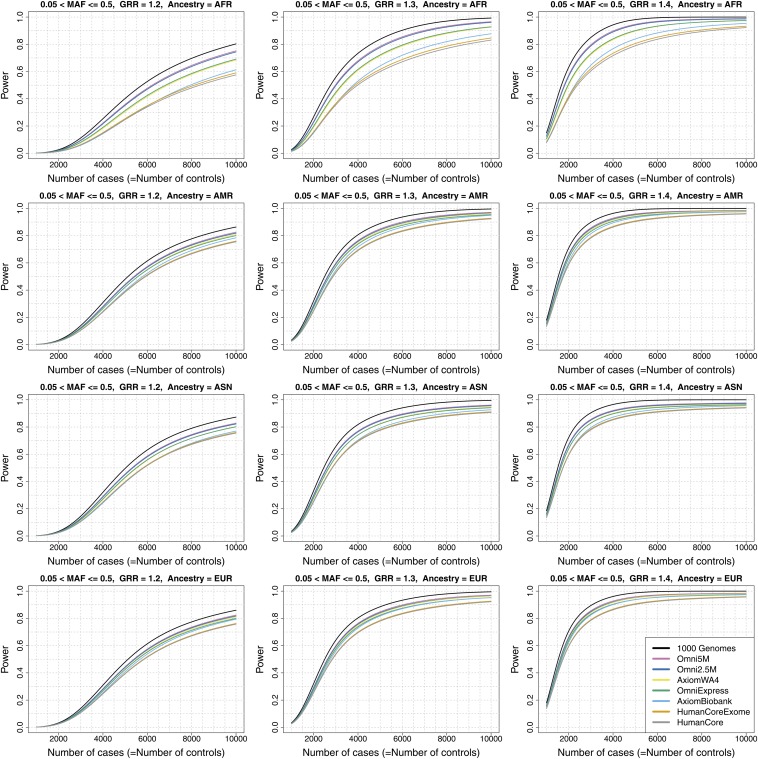
Microarray single-nucleotide polymorphism genotyping, combined with imputation of untyped variants, has been widely adopted as an efficient means to interrogate variation across the human genome. "Genomic coverage" is the total proportion of genomic variation captured by an array, either by direct observation or through an indirect means such as linkage disequilibrium or imputation. We have performed imputation-based genomic coverage assessments of eight current genotyping arrays that assay from ~0.3 to ~5 million variants. Coverage was determined separately in each of the four continental ancestry groups in the 1000 Genomes Project phase 1 release. We used the subset of 1000 Genomes variants present on each array to impute the remaining variants and assessed coverage based on correlation between imputed and observed allelic dosages. More than 75% of common variants (minor allele frequency > 0.05) are covered by all arrays in all groups except for African ancestry, and up to ~90% in all ancestries for the highest density arrays. In contrast, less than 40% of less common variants (0.01 < minor allele frequency < 0.05) are covered by low density arrays in all ancestries and 50-80% in high density arrays, depending on ancestry. We also calculated genome-wide power to detect variant-trait association in a case-control design, across varying sample sizes, effect sizes, and minor allele frequency ranges, and compare these array-based power estimates with a hypothetical array that would type all variants in 1000 Genomes. These imputation-based genomic coverage and power analyses are intended as a practical guide to researchers planning genetic studies.
微阵列单核苷酸多态性基因分型,结合未分型变异的填充,已被广泛用作探究人类基因组变异的有效手段。“基因组覆盖率”是指阵列通过直接观察或通过连锁不平衡或填充等间接手段捕获的基因组变异的总比例。我们对目前的八款基因分型阵列进行了基于填充的基因组覆盖率评估,这些阵列检测的变异数量从约30万到约500万不等。在千人基因组计划第一阶段发布的数据中,我们分别在四个大陆祖先群体中确定了覆盖率。我们使用每个阵列上存在的千人基因组变异子集来填充其余变异,并根据填充和观察到的等位基因剂量之间的相关性评估覆盖率。除非洲祖先群体外,所有群体中所有阵列都覆盖了超过75%的常见变异(次要等位基因频率>0.05),对于最高密度的阵列,所有祖先群体中的覆盖率高达约90%。相比之下,低密度阵列在所有祖先群体中覆盖的罕见变异(0.01<次要等位基因频率<0.05)不到40%,高密度阵列中的覆盖率为50-80%,具体取决于祖先群体。我们还计算了在病例对照设计中检测变异与性状关联的全基因组效能,涵盖了不同的样本量、效应大小和次要等位基因频率范围,并将这些基于阵列的效能估计与一个假设的阵列进行比较,该假设阵列将对千人基因组中的所有变异进行分型。这些基于填充的基因组覆盖率和效能分析旨在为计划进行基因研究的研究人员提供实用指南。