Department of Computer Science, University of California, Los Angeles, CA 90024, USA.
Bioinformatics. 2011 Jul 1;27(13):i288-94. doi: 10.1093/bioinformatics/btr221.
The analysis of gene coexpression is at the core of many types of genetic analysis. The coexpression between two genes can be calculated by using a traditional Pearson's correlation coefficient. However, unobserved confounding effects may cause inflation of the Pearson's correlation so that uncorrelated genes appear correlated. Many general methods have been suggested, which aim to remove the effects of confounding from gene expression data. However, the residual confounding which is not accounted for by these generic correction procedures has the potential to induce correlation between genes. Therefore, a method that specifically aims to calculate gene coexpression between gene expression arrays, while accounting for confounding effects, is desirable.
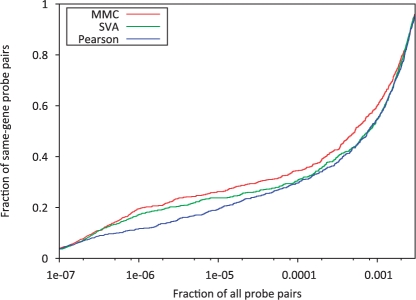
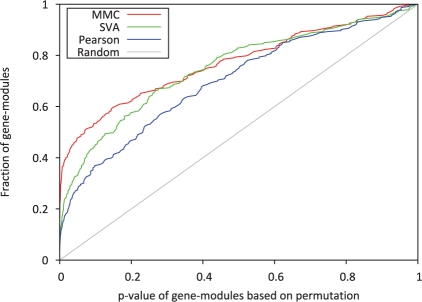
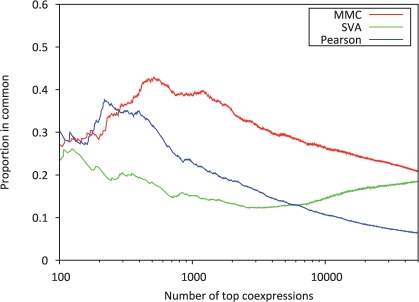
In this article, we present a statistical model for calculating gene coexpression called mixed model coexpression (MMC), which models coexpression within a mixed model framework. Confounding effects are expected to be encoded in the matrix representing the correlation between arrays, the inter-sample correlation matrix. By conditioning on the information in the inter-sample correlation matrix, MMC is able to produce gene coexpressions that are not influenced by global confounding effects and thus significantly reduce the number of spurious coexpressions observed. We applied MMC to both human and yeast datasets and show it is better able to effectively prioritize strong coexpressions when compared to a traditional Pearson's correlation and a Pearson's correlation applied to data corrected with surrogate variable analysis (SVA).
The method is implemented in the R programming language and may be found at http://genetics.cs.ucla.edu/mmc.
基因共表达分析是许多类型的遗传分析的核心。两个基因之间的共表达可以使用传统的皮尔逊相关系数来计算。然而,未观察到的混杂效应可能会导致皮尔逊相关系数的膨胀,从而使不相关的基因表现出相关性。已经提出了许多通用方法,旨在从基因表达数据中去除混杂效应的影响。然而,这些通用校正程序未考虑到的残留混杂因素有可能导致基因之间的相关性。因此,需要一种专门旨在计算基因表达阵列之间基因共表达的方法,同时考虑混杂效应。
在本文中,我们提出了一种称为混合模型共表达(MMC)的计算基因共表达的统计模型,该模型在混合模型框架内对共表达进行建模。混杂效应预计将被编码在表示阵列之间相关性的矩阵中,即样本间相关矩阵中。通过对样本间相关矩阵中的信息进行条件化,MMC 能够产生不受全局混杂效应影响的基因共表达,从而显著减少观察到的虚假共表达数量。我们将 MMC 应用于人类和酵母数据集,并表明与传统的皮尔逊相关系数和应用于替代变量分析(SVA)校正后数据的皮尔逊相关系数相比,它能够更有效地优先考虑强共表达。
该方法在 R 编程语言中实现,可在 http://genetics.cs.ucla.edu/mmc 找到。