School of Mathematical Sciences, Tel Aviv University, Tel Aviv, Israel.
Bioinformatics. 2011 Jul 1;27(13):i317-23. doi: 10.1093/bioinformatics/btr219.
Random effects models have recently been introduced as an approach for analyzing genome wide association studies (GWASs), which allows estimation of overall heritability of traits without explicitly identifying the genetic loci responsible. Using this approach, Yang et al. (2010) have demonstrated that the heritability of height is much higher than the ~10% associated with identified genetic factors. However, Yang et al. (2010) relied on a heuristic for performing estimation in this model.
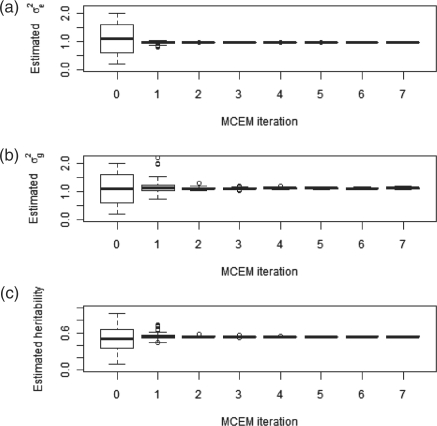
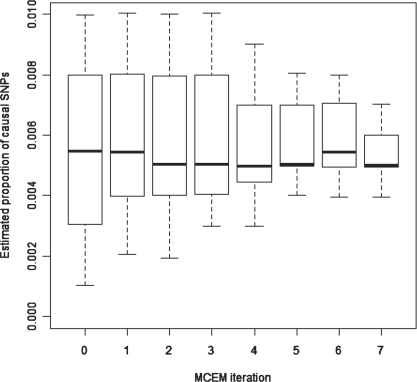
We adopt the model framework of Yang et al. (2010) and develop a method for maximum-likelihood (ML) estimation in this framework. Our method is based on Monte-Carlo expectation-maximization (MCEM; Wei et al., 1990), an expectation-maximization algorithm wherein a Markov chain Monte Carlo approach is used in the E-step. We demonstrate that this method leads to more stable and accurate heritability estimation compared to the approach of Yang et al. (2010), and it also allows us to find ML estimates of the portion of markers which are causal, indicating whether the heritability stems from a small number of powerful genetic factors or a large number of less powerful ones.
随机效应模型最近被引入作为分析全基因组关联研究(GWAS)的一种方法,它允许在不明确识别负责的遗传基因座的情况下估计性状的总体遗传率。Yang 等人(2010 年)使用这种方法表明,身高的遗传率远高于与已确定的遗传因素相关的约 10%。然而,Yang 等人(2010 年)在该模型中依赖启发式方法进行估计。
我们采用了 Yang 等人(2010 年)的模型框架,并为该框架开发了一种最大似然(ML)估计方法。我们的方法基于蒙特卡罗期望最大化(MCEM;Wei 等人,1990 年),这是一种期望最大化算法,其中在 E 步中使用马尔可夫链蒙特卡罗方法。我们证明与 Yang 等人(2010 年)的方法相比,这种方法导致更稳定和准确的遗传率估计,并且它还允许我们找到因果标记的 ML 估计,表明遗传率是源于少数强大的遗传因素还是大量较弱的遗传因素。