Bioinformatics Core Facility, Department of Molecular Medicine, Beckman Research Institute, City of Hope Medical Center, Duarte, CA 91010, USA.
BMC Bioinformatics. 2011 Jun 29;12:267. doi: 10.1186/1471-2105-12-267.
The popularity of massively parallel exome and transcriptome sequencing projects demands new data mining tools with a comprehensive set of features to support a wide range of analysis tasks.
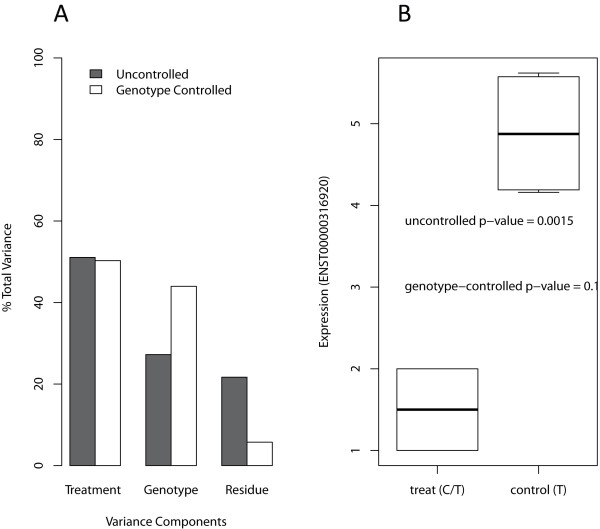
SeqGene, a new data mining tool, supports mutation detection and annotation, dbSNP and 1000 Genome data integration, RNA-Seq expression quantification, mutation and coverage visualization, allele specific expression (ASE), differentially expressed genes (DEGs) identification, copy number variation (CNV) analysis, and gene expression quantitative trait loci (eQTLs) detection. We also developed novel methods for testing the association between SNP and expression and identifying genotype-controlled DEGs. We showed that the results generated from SeqGene compares favourably to other existing methods in our case studies.
SeqGene is designed as a general-purpose software package. It supports both paired-end reads and single reads generated on most sequencing platforms; it runs on all major types of computers; it supports arbitrary genome assemblies for arbitrary organisms; and it scales well to support both large and small scale sequencing projects. The software homepage is http://seqgene.sourceforge.net.
高通量外显子组和转录组测序项目的普及,需要新的数据挖掘工具,这些工具应具有全面的功能集,以支持广泛的分析任务。
SeqGene 是一个新的数据挖掘工具,支持突变检测和注释、dbSNP 和 1000 基因组数据集成、RNA-Seq 表达定量、突变和覆盖可视化、等位基因特异性表达(ASE)、差异表达基因(DEGs)鉴定、拷贝数变异(CNV)分析和基因表达数量性状位点(eQTLs)检测。我们还开发了用于测试 SNP 与表达之间关联和鉴定基因型控制的 DEGs 的新方法。我们表明,在我们的案例研究中,SeqGene 生成的结果优于其他现有方法。
SeqGene 被设计为通用软件包。它支持大多数测序平台生成的配对末端读取和单端读取;它在所有主要类型的计算机上运行;它支持任意生物体的任意基因组组装;并且它可以很好地扩展以支持大规模和小规模测序项目。软件主页是 http://seqgene.sourceforge.net。