Günther Torsten, Schmid Karl J
Institute of Plant Breeding, Seed Science and Population Genetics, University of Hohenheim, Stuttgart, Germany.
BMC Res Notes. 2011 Jul 5;4:232. doi: 10.1186/1756-0500-4-232.
The increasing amount of genome information allows us to address various questions regarding the molecular evolution and population genetics of different species. Such genome-wide data sets including thousands of individuals genotyped at hundreds of thousands of markers require time-efficient and powerful analysis methods. Demography and sampling introduce a bias into present population genetic tests of natural selection, which may confound results. Thus, a modification of test statistics is necessary to introduce time-efficient and unbiased analysis methods.
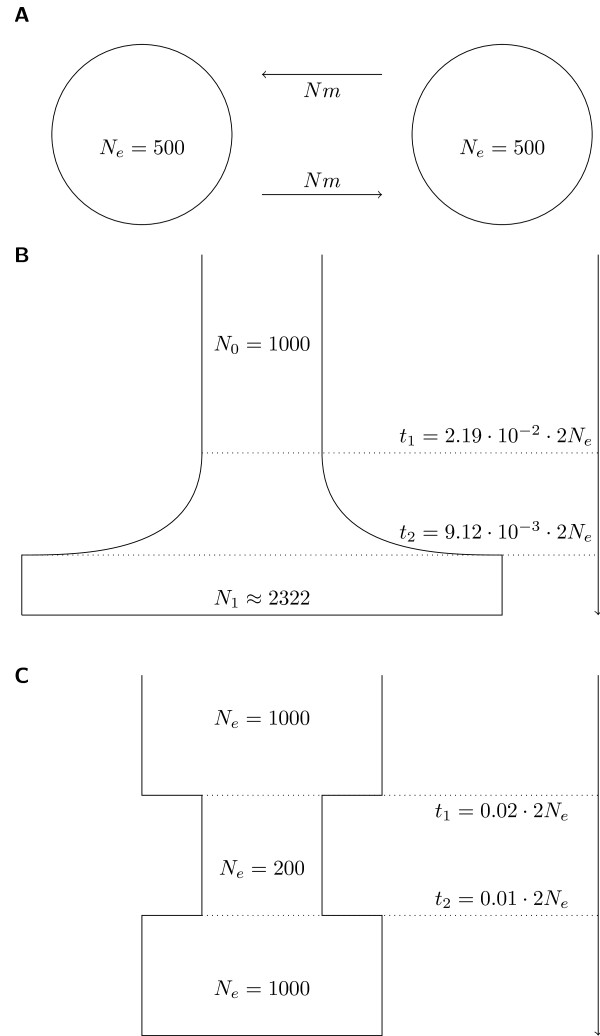
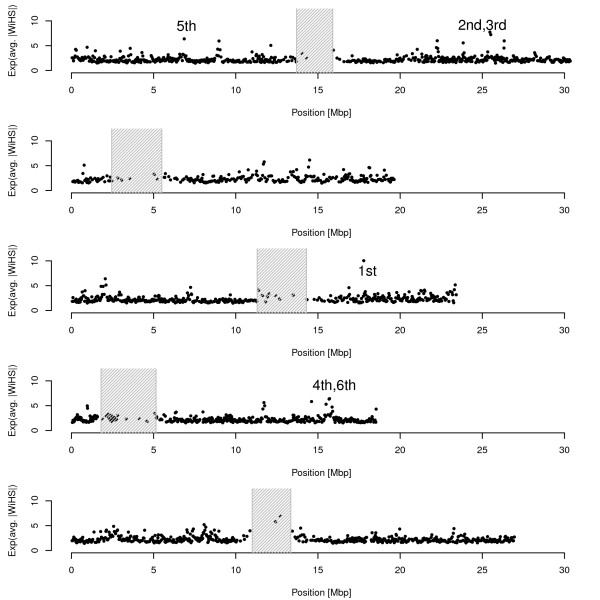
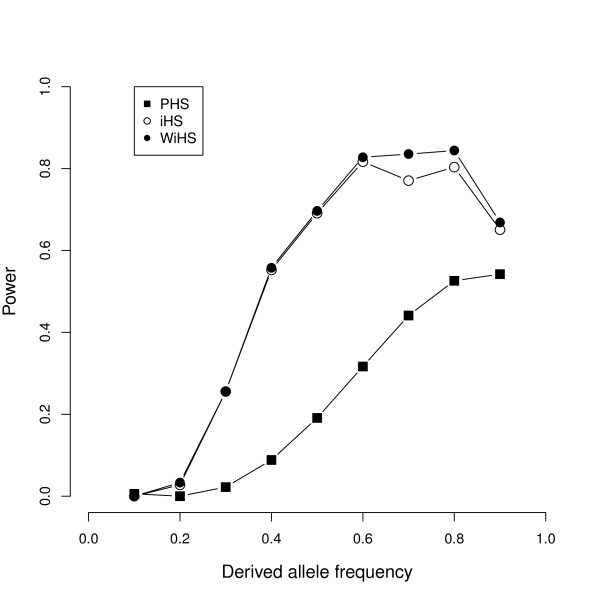
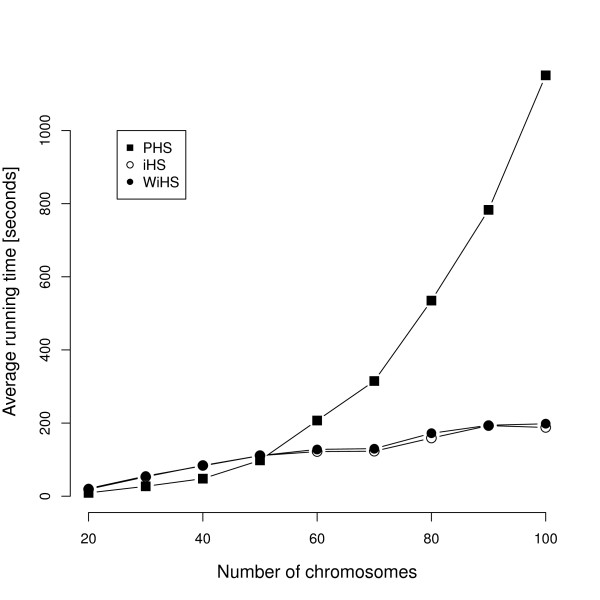
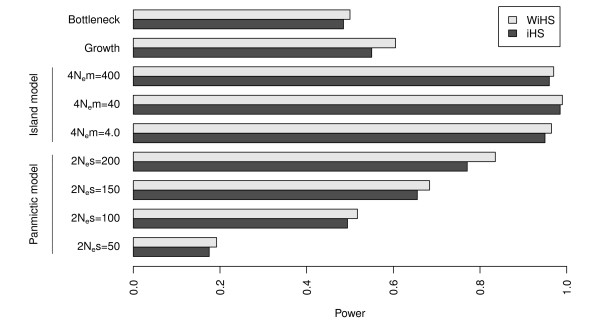
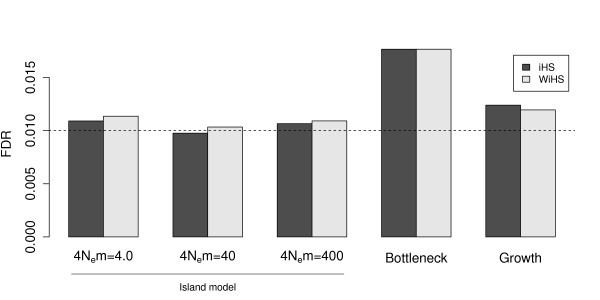
We present an improved haplotype-based test of selective sweeps in samples of unequally related individuals. For this purpose, we modified existing tests by weighting the contribution of each individual based on its uniqueness in the entire sample. In contrast to previous tests, this modified test is feasible even for large genome-wide data sets of multiple individuals. We utilize coalescent simulations to estimate the sensitivity of such haplotype-based test statistics to complex demographic scenarios, such as population structure, population growth and bottlenecks. The analysis of empirical data from humans reveals different results compared to previous tests. Additionally, we show that our statistic is applicable to empirical data from Arabidopsis thaliana. Overall, the modified test leads to a slight but significant increase of power to detect selective sweeps among all demographic scenarios.
The concept of this modification might be applied to other statistics in population genetics to reduce the intrinsic bias of demography and sampling. Additionally, the combination of different test statistics may further improve the performance of tests for natural selection.
基因组信息数量的不断增加使我们能够解决有关不同物种分子进化和群体遗传学的各种问题。此类全基因组数据集包含成千上万个个体在成百上千个标记上的基因型信息,需要高效且强大的分析方法。群体统计学和抽样会给当前自然选择的群体遗传学检测带来偏差,这可能会混淆结果。因此,有必要对检验统计量进行修正,以引入高效且无偏差的分析方法。
我们提出了一种改进的基于单倍型的方法,用于检测非等亲缘关系个体样本中的选择性清除。为此,我们通过根据每个个体在整个样本中的独特性对其贡献进行加权,对现有检验方法进行了修正。与之前的检验不同,这种修正后的检验即使对于多个个体的大型全基因组数据集也是可行的。我们利用合并模拟来估计这种基于单倍型的检验统计量对复杂群体统计学情景(如群体结构、群体增长和瓶颈效应)的敏感性。对人类经验数据的分析显示,与之前的检验相比,结果有所不同。此外,我们表明我们的统计量适用于拟南芥的经验数据。总体而言,修正后的检验在所有群体统计学情景下检测选择性清除的能力略有但显著提高。
这种修正的概念可能适用于群体遗传学中的其他统计量,以减少群体统计学和抽样的内在偏差。此外,不同检验统计量的组合可能会进一步提高自然选择检验的性能。