Prussia Andrew, Thepchatri Pahk, Snyder James P, Plemper Richard K
Department of Chemistry, Emory University, Atlanta, GA 30322, USA; E-Mails:
Int J Mol Sci. 2011;12(6):4027-52. doi: 10.3390/ijms12064027. Epub 2011 Jun 15.
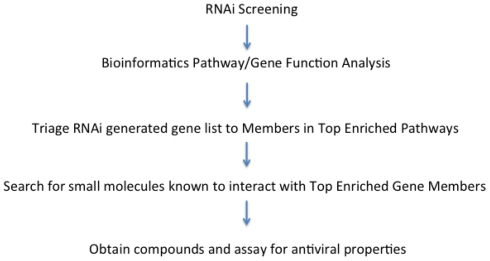
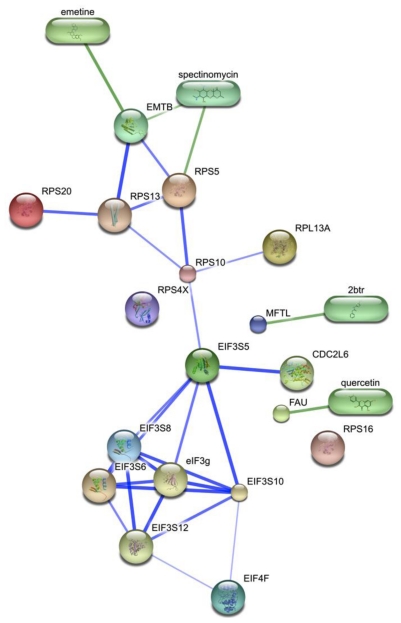
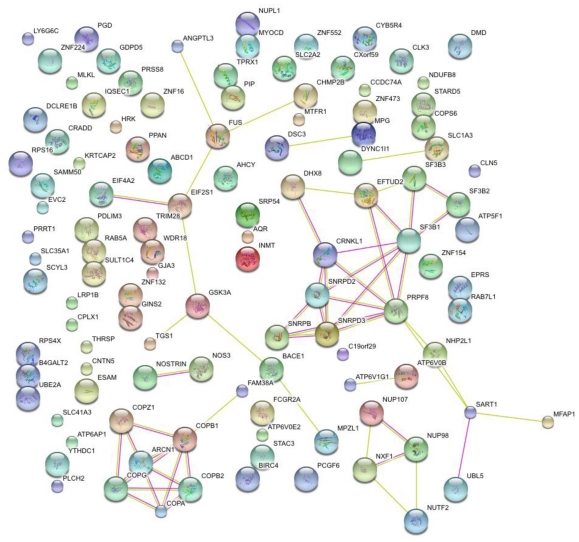
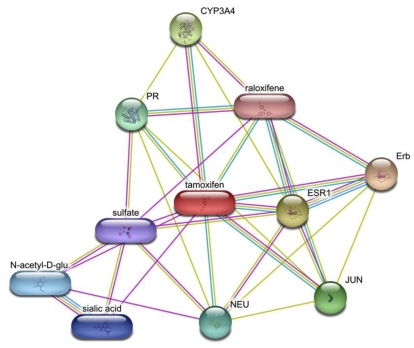
Since the onset of antiviral therapy, viral resistance has compromised the clinical value of small-molecule drugs targeting pathogen components. As intracellular parasites, viruses complete their life cycle by hijacking a multitude of host-factors. Aiming at the latter rather than the pathogen directly, host-directed antiviral therapy has emerged as a concept to counteract evolution of viral resistance and develop broad-spectrum drug classes. This approach is propelled by bioinformatics analysis of genome-wide screens that greatly enhance insights into the complex network of host-pathogen interactions and generate a shortlist of potential gene targets from a multitude of candidates, thus setting the stage for a new era of rational identification of drug targets for host-directed antiviral therapies. With particular emphasis on human immunodeficiency virus and influenza virus, two major human pathogens, we review screens employed to elucidate host-pathogen interactions and discuss the state of database ontology approaches applicable to defining a therapeutic endpoint. The value of this strategy for drug discovery is evaluated, and perspectives for bioinformatics-driven hit identification are outlined.
自抗病毒治疗开始以来,病毒耐药性损害了针对病原体成分的小分子药物的临床价值。作为细胞内寄生虫,病毒通过劫持多种宿主因子来完成其生命周期。宿主导向的抗病毒治疗不是直接针对病原体,而是针对宿主因子,作为一种对抗病毒耐药性进化和开发广谱药物类别的概念应运而生。全基因组筛选的生物信息学分析推动了这种方法的发展,该分析极大地增强了对宿主-病原体相互作用复杂网络的认识,并从众多候选物中生成潜在基因靶点的清单,从而为合理识别宿主导向的抗病毒治疗药物靶点的新时代奠定了基础。我们特别强调两种主要的人类病原体——人类免疫缺陷病毒和流感病毒,回顾用于阐明宿主-病原体相互作用的筛选方法,并讨论适用于定义治疗终点的数据库本体方法的现状。评估了该策略在药物发现中的价值,并概述了生物信息学驱动的命中识别的前景。