Department of Biological Engineering, Massachusetts Institute of Technology, Cambridge, Massachusetts, USA.
PLoS Comput Biol. 2011 Jul;7(7):e1002119. doi: 10.1371/journal.pcbi.1002119. Epub 2011 Jul 21.
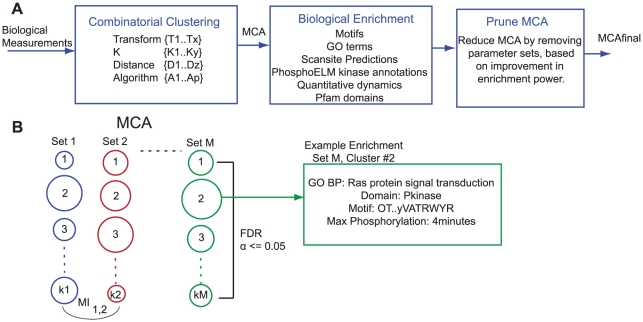
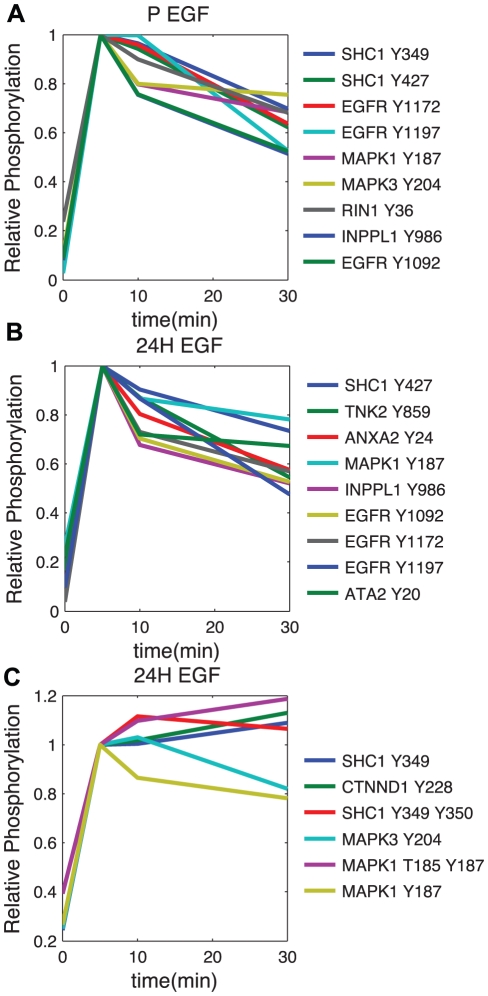
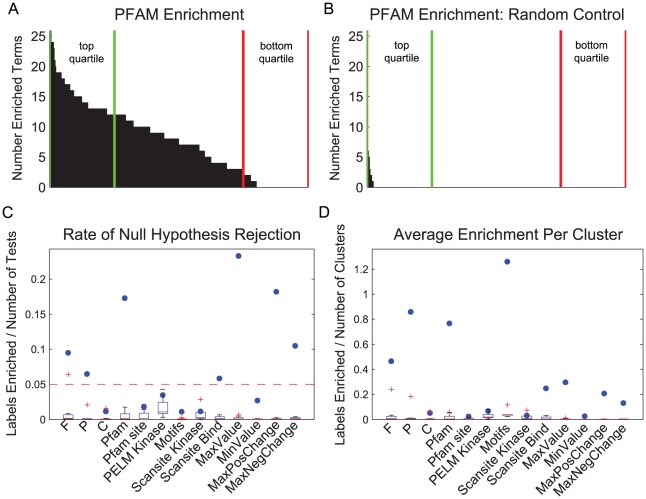
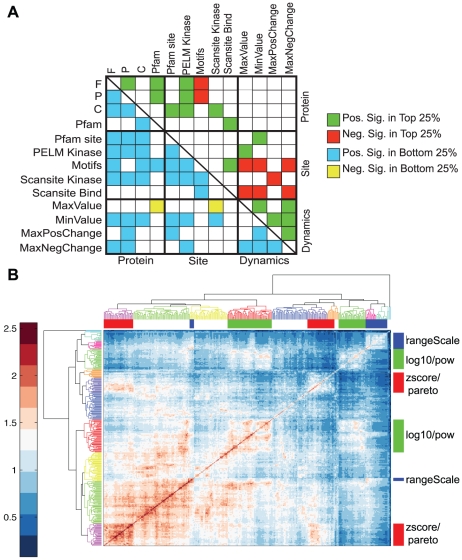
Advances in proteomic technologies continue to substantially accelerate capability for generating experimental data on protein levels, states, and activities in biological samples. For example, studies on receptor tyrosine kinase signaling networks can now capture the phosphorylation state of hundreds to thousands of proteins across multiple conditions. However, little is known about the function of many of these protein modifications, or the enzymes responsible for modifying them. To address this challenge, we have developed an approach that enhances the power of clustering techniques to infer functional and regulatory meaning of protein states in cell signaling networks. We have created a new computational framework for applying clustering to biological data in order to overcome the typical dependence on specific a priori assumptions and expert knowledge concerning the technical aspects of clustering. Multiple clustering analysis methodology ('MCAM') employs an array of diverse data transformations, distance metrics, set sizes, and clustering algorithms, in a combinatorial fashion, to create a suite of clustering sets. These sets are then evaluated based on their ability to produce biological insights through statistical enrichment of metadata relating to knowledge concerning protein functions, kinase substrates, and sequence motifs. We applied MCAM to a set of dynamic phosphorylation measurements of the ERRB network to explore the relationships between algorithmic parameters and the biological meaning that could be inferred and report on interesting biological predictions. Further, we applied MCAM to multiple phosphoproteomic datasets for the ERBB network, which allowed us to compare independent and incomplete overlapping measurements of phosphorylation sites in the network. We report specific and global differences of the ERBB network stimulated with different ligands and with changes in HER2 expression. Overall, we offer MCAM as a broadly-applicable approach for analysis of proteomic data which may help increase the current understanding of molecular networks in a variety of biological problems.
蛋白质组学技术的进步继续极大地提高了在生物样本中生成蛋白质水平、状态和活性的实验数据的能力。例如,现在可以研究受体酪氨酸激酶信号网络,以捕获多种条件下数百到数千种蛋白质的磷酸化状态。然而,对于许多这些蛋白质修饰的功能,或者负责修饰它们的酶,人们知之甚少。为了应对这一挑战,我们开发了一种方法,该方法增强了聚类技术推断细胞信号网络中蛋白质状态的功能和调节意义的能力。我们创建了一个新的计算框架,用于将聚类应用于生物数据,以克服对聚类技术特定先验假设和专家知识的典型依赖。多聚类分析方法('MCAM')以组合方式使用一系列不同的数据转换、距离度量、集合大小和聚类算法,来创建一组聚类集。然后,根据它们通过与蛋白质功能、激酶底物和序列基序相关的元数据的统计富集来产生生物学见解的能力来评估这些集。我们将 MCAM 应用于 ERRB 网络的一组动态磷酸化测量中,以探索算法参数与可以推断和报告有趣生物学预测的生物学意义之间的关系。此外,我们将 MCAM 应用于 ERBB 网络的多个磷酸蛋白质组学数据集,这使我们能够比较网络中磷酸化位点的独立和不完整重叠测量。我们报告了不同配体和 HER2 表达变化刺激下 ERBB 网络的特定和全局差异。总体而言,我们提供了 MCAM 作为一种广泛适用的蛋白质组学数据分析方法,这可能有助于提高对各种生物学问题中分子网络的当前理解。