Pacific Northwest National Laboratory, Richland, WA 99352, USA.
Bioinformatics. 2011 Oct 15;27(20):2866-72. doi: 10.1093/bioinformatics/btr479. Epub 2011 Aug 18.
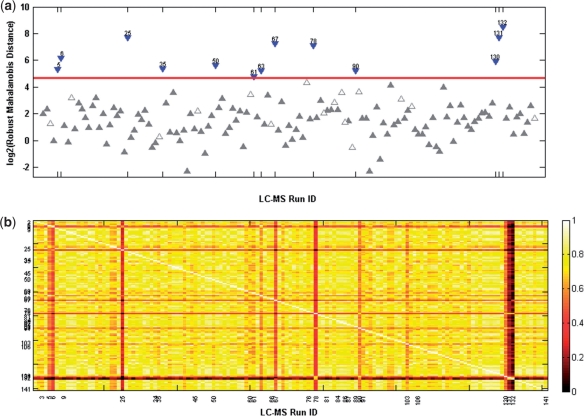
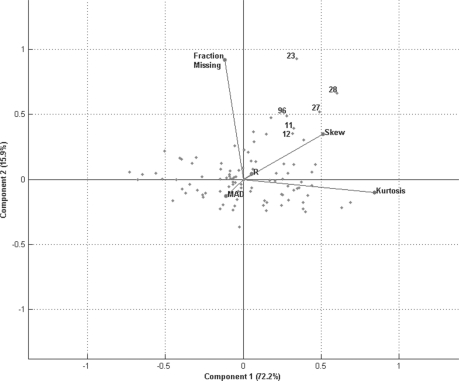
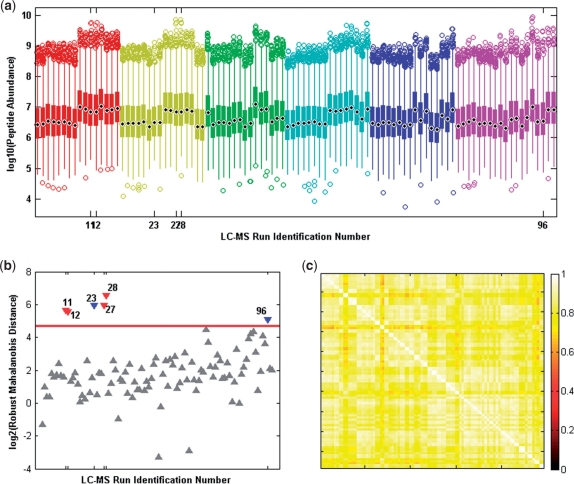
In the analysis of differential peptide peak intensities (i.e. abundance measures), LC-MS analyses with poor quality peptide abundance data can bias downstream statistical analyses and hence the biological interpretation for an otherwise high-quality dataset. Although considerable effort has been placed on assuring the quality of the peptide identification with respect to spectral processing, to date quality assessment of the subsequent peptide abundance data matrix has been limited to a subjective visual inspection of run-by-run correlation or individual peptide components. Identifying statistical outliers is a critical step in the processing of proteomics data as many of the downstream statistical analyses [e.g. analysis of variance (ANOVA)] rely upon accurate estimates of sample variance, and their results are influenced by extreme values.
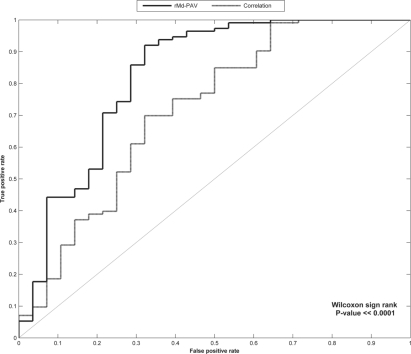
We describe a novel multivariate statistical strategy for the identification of LC-MS runs with extreme peptide abundance distributions. Comparison with current method (run-by-run correlation) demonstrates a significantly better rate of identification of outlier runs by the multivariate strategy. Simulation studies also suggest that this strategy significantly outperforms correlation alone in the identification of statistically extreme liquid chromatography-mass spectrometry (LC-MS) runs.
https://www.biopilot.org/docs/Software/RMD.php
Supplementary material is available at Bioinformatics online.
在分析差异肽峰强度(即丰度测量)时,LC-MS 分析中肽丰度数据质量较差会偏向下游统计分析,从而影响原本高质量数据集的生物学解释。尽管已经在保证肽鉴定质量方面付出了相当大的努力,但是到目前为止,对后续肽丰度数据矩阵的质量评估仅限于对逐个运行的相关性或单个肽成分的主观视觉检查。在处理蛋白质组学数据时,识别统计异常值是一个关键步骤,因为许多下游统计分析(例如方差分析(ANOVA))都依赖于对样本方差的准确估计,并且它们的结果受到极值的影响。
我们描述了一种用于识别具有极端肽丰度分布的 LC-MS 运行的新的多变量统计策略。与当前方法(运行间相关性)的比较表明,多变量策略显著提高了异常运行的识别率。模拟研究还表明,该策略在识别统计学上极端的液相色谱-质谱(LC-MS)运行方面明显优于仅相关性。
https://www.biopilot.org/docs/Software/RMD.php
补充材料可在 Bioinformatics 在线获取。