Department of Pharmacology and Chemical Biology, University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania, United States of America.
PLoS One. 2011;6(10):e25823. doi: 10.1371/journal.pone.0025823. Epub 2011 Oct 3.
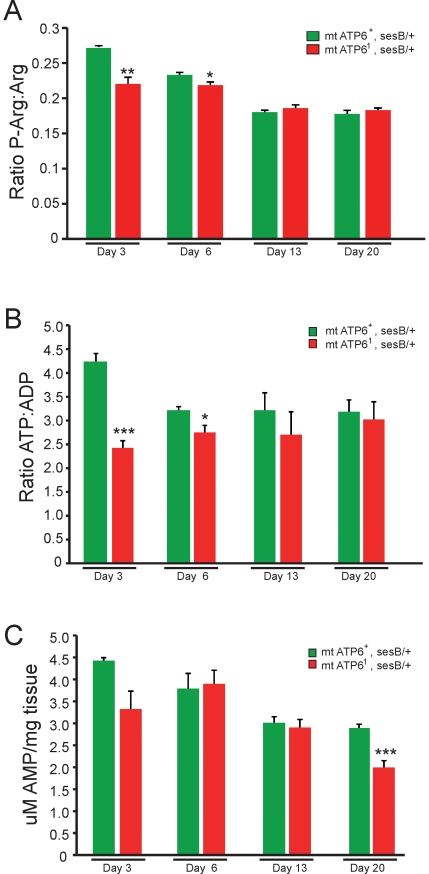
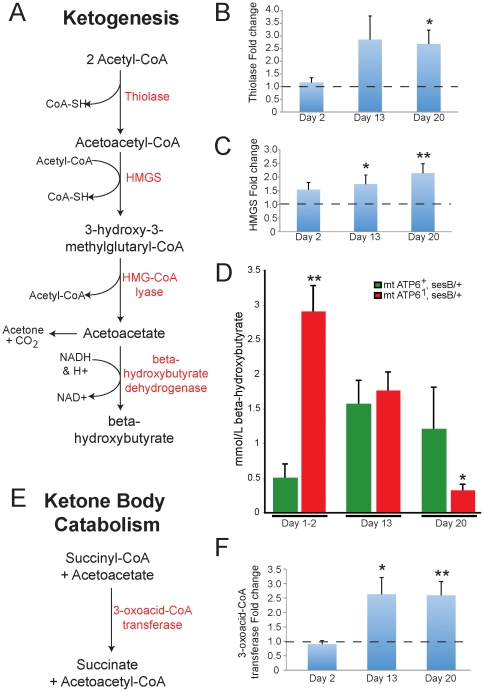
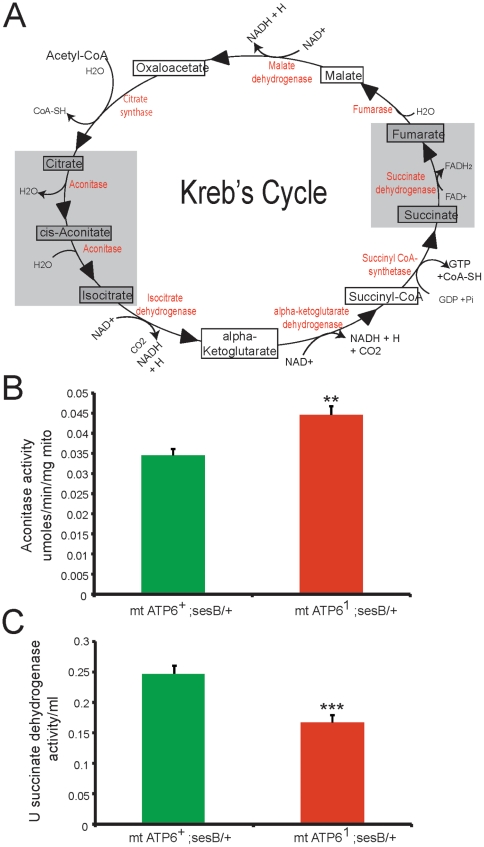
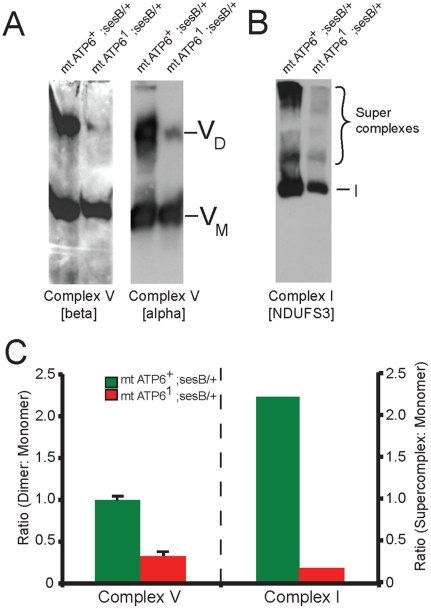
Numerous mitochondrial DNA mutations cause mitochondrial encephalomyopathy: a collection of related diseases for which there exists no effective treatment. Mitochondrial encephalomyopathies are complex multisystem diseases that exhibit a relentless progression of severity, making them both difficult to treat and study. The pathogenic and compensatory metabolic changes that are associated with chronic mitochondrial dysfunction are not well understood. The Drosophila ATP6(1) mutant models human mitochondrial encephalomyopathy and allows the study of metabolic changes and compensation that occur throughout the lifetime of an affected animal. ATP6(1)animals have a nearly complete loss of ATP synthase activity and an acute bioenergetic deficit when they are asymptomatic, but surprisingly we discovered no chronic bioenergetic deficit in these animals during their symptomatic period. Our data demonstrate dynamic metabolic compensatory mechanisms that sustain normal energy availability and activity despite chronic mitochondrial complex V dysfunction resulting from an endogenous mutation in the mitochondrial DNA. ATP6(1)animals compensate for their loss of oxidative phosphorylation through increases in glycolytic flux, ketogenesis and Kreb's cycle activity early during pathogenesis. However, succinate dehydrogenase activity is reduced and mitochondrial supercomplex formation is severely disrupted contributing to the pathogenesis seen in ATP6(1) animals. These studies demonstrate the dynamic nature of metabolic compensatory mechanisms and emphasize the need for time course studies in tractable animal systems to elucidate disease pathogenesis and novel therapeutic avenues.
许多线粒体 DNA 突变导致线粒体脑肌病:这是一组相关疾病,目前尚无有效的治疗方法。线粒体脑肌病是一种复杂的多系统疾病,其严重程度不断恶化,因此既难以治疗也难以研究。与慢性线粒体功能障碍相关的致病和代偿代谢变化尚不清楚。果蝇 ATP6(1)突变模型模拟了人类线粒体脑肌病,使我们能够研究在受影响动物的整个生命周期中发生的代谢变化和代偿。ATP6(1)动物在无症状时几乎完全丧失 ATP 合酶活性,并且存在急性生物能量缺陷,但令人惊讶的是,我们在这些动物出现症状期间并未发现慢性生物能量缺陷。我们的数据表明,尽管由于线粒体 DNA 中的内源性突变导致线粒体复合物 V 功能持续慢性障碍,但存在动态代谢代偿机制,可维持正常的能量供应和活性。ATP6(1)动物通过在发病早期增加糖酵解通量、酮生成和克雷布斯循环活性来补偿氧化磷酸化的丧失。然而,琥珀酸脱氢酶活性降低,线粒体超复合体形成严重受损,这导致了 ATP6(1)动物中观察到的发病机制。这些研究表明了代谢代偿机制的动态性质,并强调了在可处理的动物系统中进行时程研究以阐明疾病发病机制和新的治疗途径的必要性。