Department of Chemistry and Chemical Biology, BioMaPS Institute for Quantitative Biology, Rutgers, The State University of New Jersey, Piscataway, New Jersey 08854, USA.
Proteins. 2012 Jan;80(1):111-25. doi: 10.1002/prot.23168. Epub 2011 Oct 31.
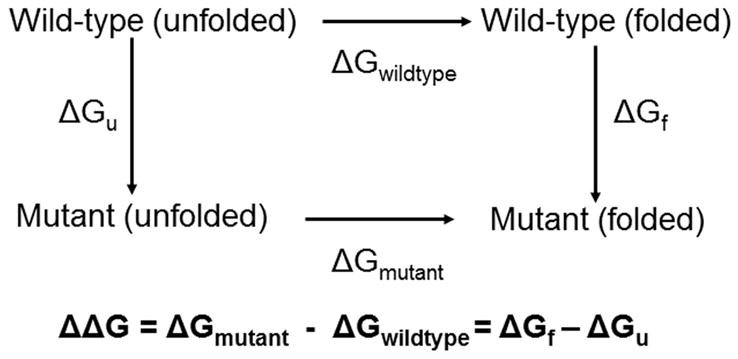
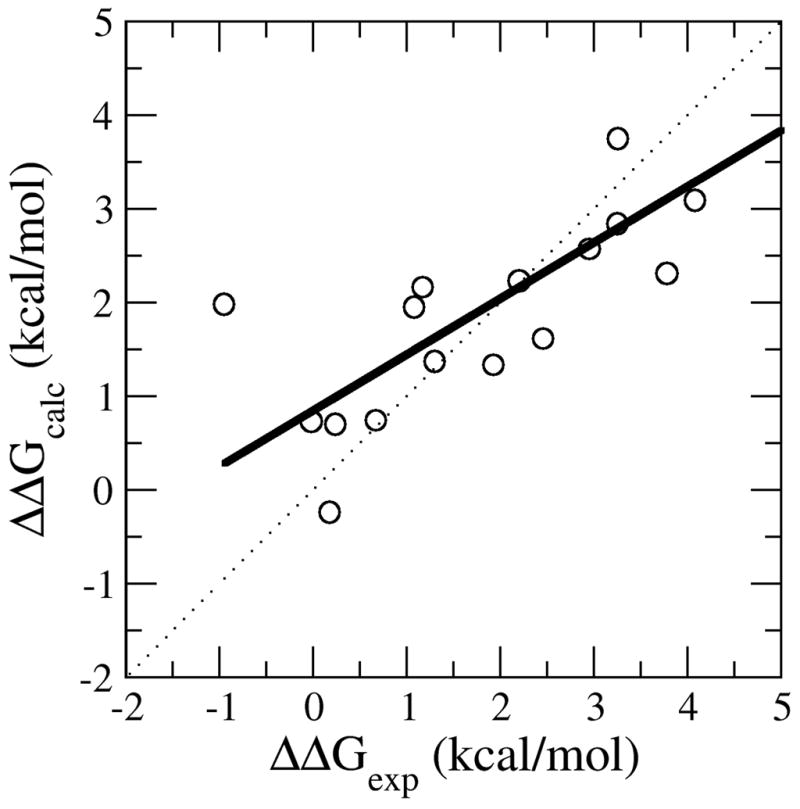
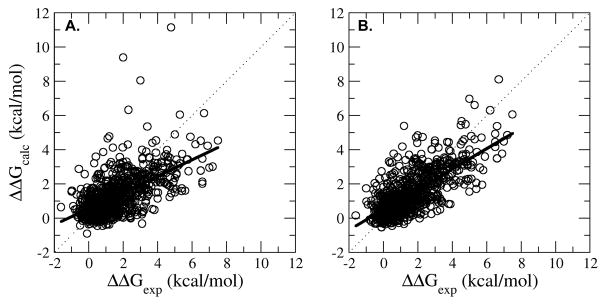
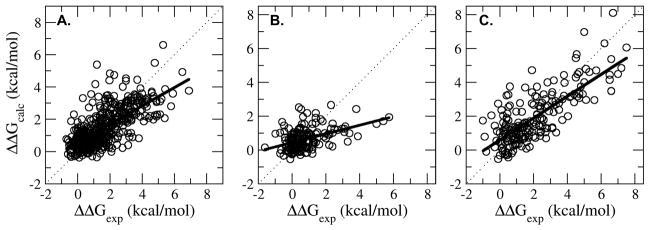
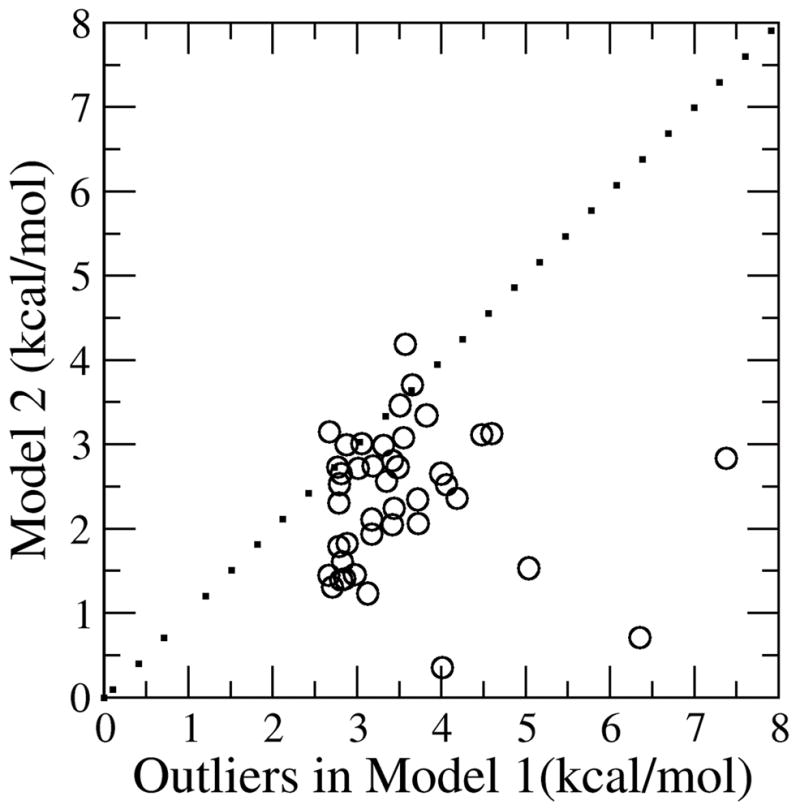
The coupling of protein energetics and sequence changes is a critical aspect of computational protein design, as well as for the understanding of protein evolution, human disease, and drug resistance. To study the molecular basis for this coupling, computational tools must be sufficiently accurate and computationally inexpensive enough to handle large amounts of sequence data. We have developed a computational approach based on the linear interaction energy (LIE) approximation to predict the changes in the free-energy of the native state induced by a single mutation. This approach was applied to a set of 822 mutations in 10 proteins which resulted in an average unsigned error of 0.82 kcal/mol and a correlation coefficient of 0.72 between the calculated and experimental ΔΔG values. The method is able to accurately identify destabilizing hot spot mutations; however, it has difficulty in distinguishing between stabilizing and destabilizing mutations because of the distribution of stability changes for the set of mutations used to parameterize the model. In addition, the model also performs quite well in initial tests on a small set of double mutations. On the basis of these promising results, we can begin to examine the relationship between protein stability and fitness, correlated mutations, and drug resistance.
蛋白质能量学和序列变化的结合是计算蛋白质设计以及理解蛋白质进化、人类疾病和耐药性的关键方面。为了研究这种结合的分子基础,计算工具必须足够准确且计算成本低廉,以处理大量的序列数据。我们开发了一种基于线性相互作用能(LIE)近似的计算方法,用于预测单个突变引起的天然状态自由能变化。该方法应用于 10 个蛋白质中的 822 个突变,导致计算和实验ΔΔG 值之间的平均无符号误差为 0.82 kcal/mol,相关系数为 0.72。该方法能够准确识别不稳定热点突变;然而,由于用于参数化模型的突变集的稳定性变化分布,它难以区分稳定和不稳定的突变。此外,该模型在一组小的双突变的初步测试中表现也相当出色。基于这些有希望的结果,我们可以开始研究蛋白质稳定性和适应性、相关突变和耐药性之间的关系。