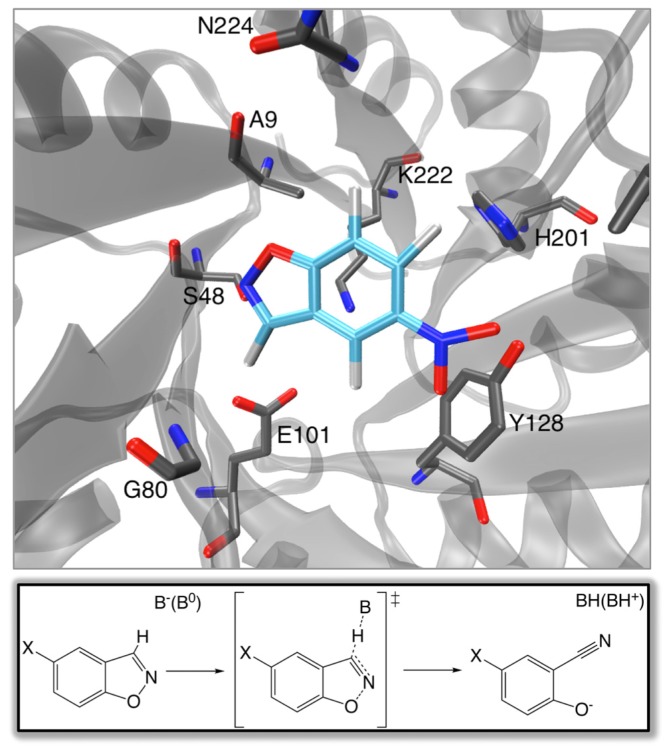
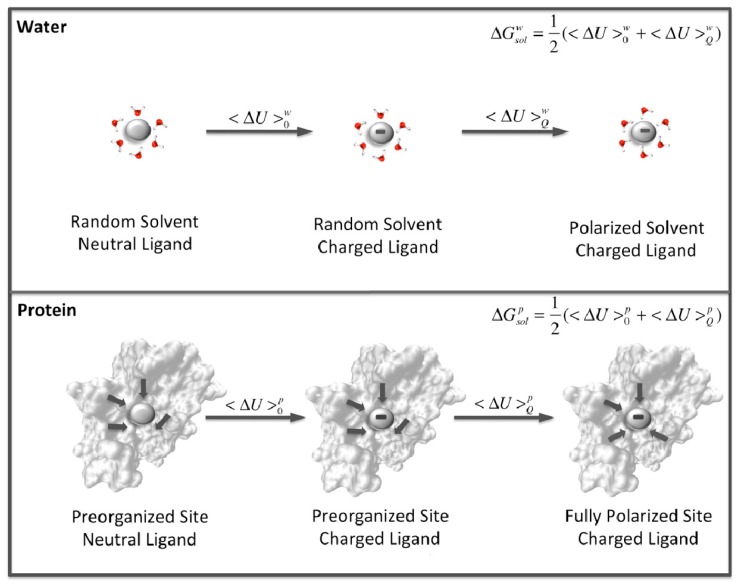
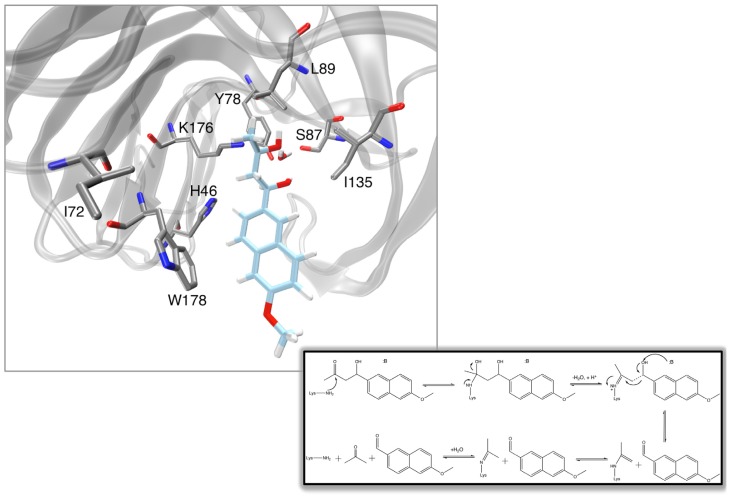
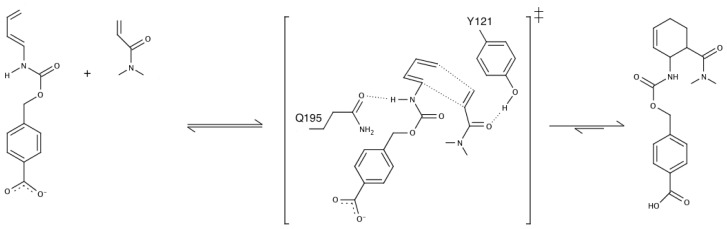
Barrozo Alexandre, Borstnar Rok, Marloie Gaël, Kamerlin Shina Caroline Lynn
Department of Cell and Molecular Biology, Uppsala Biomedical Center (BMC), Uppsala University, Box 596, S-751 24 Uppsala, Sweden.
Int J Mol Sci. 2012 Sep 28;13(10):12428-60. doi: 10.3390/ijms131012428.
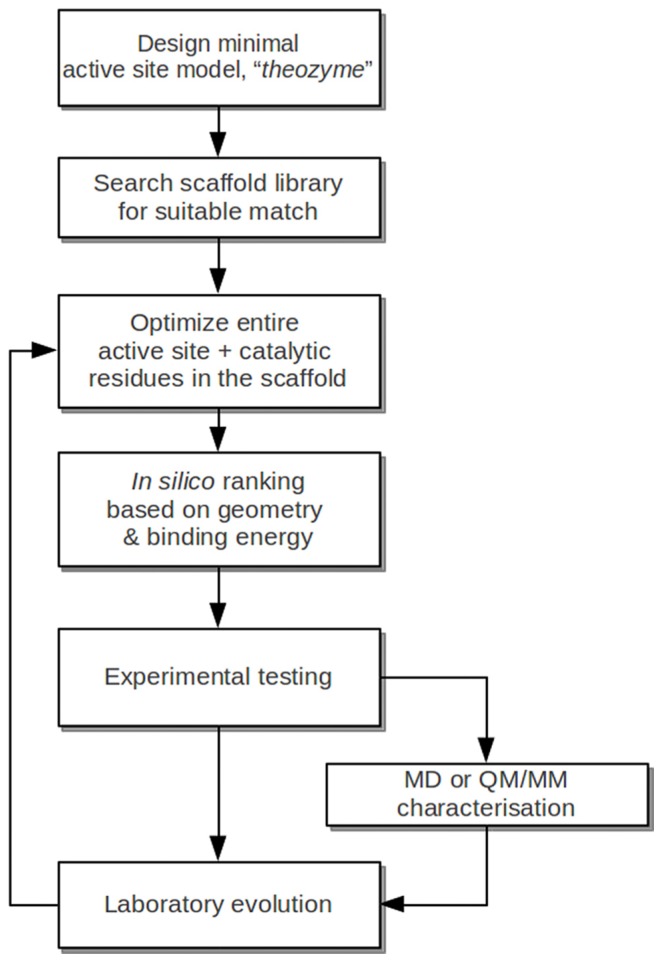
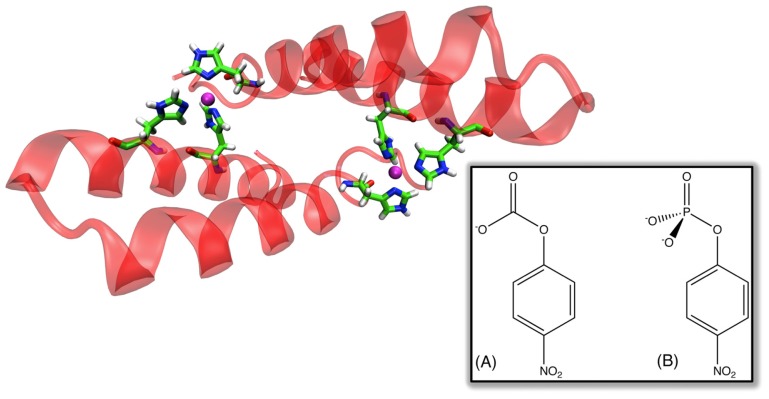
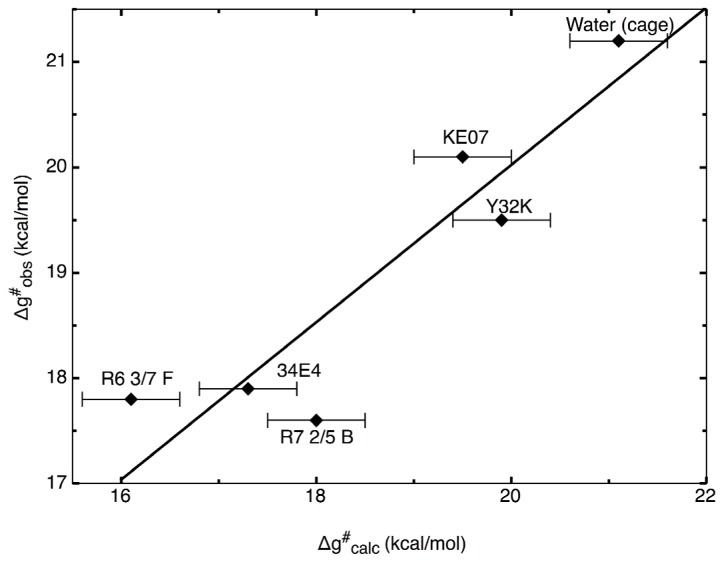
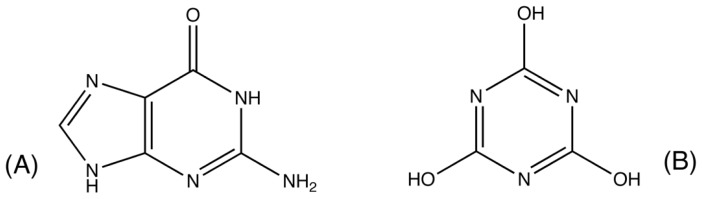
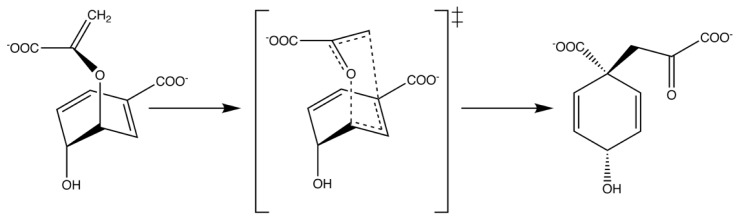
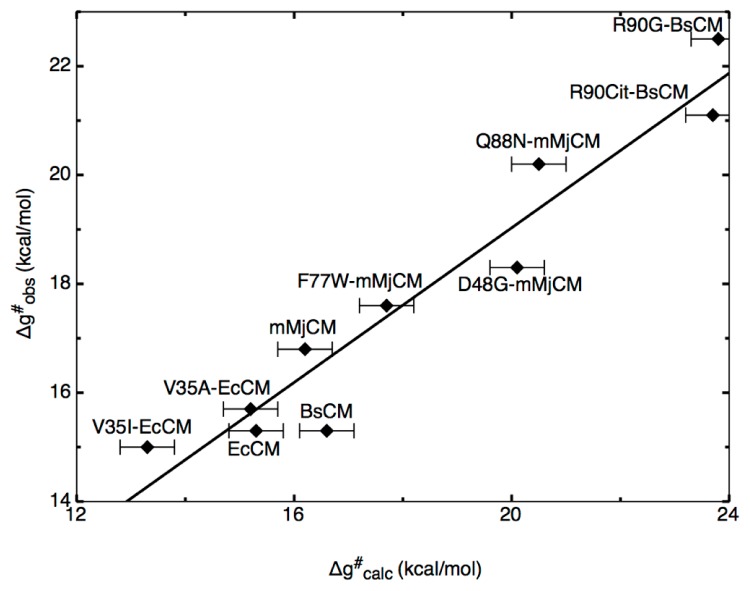
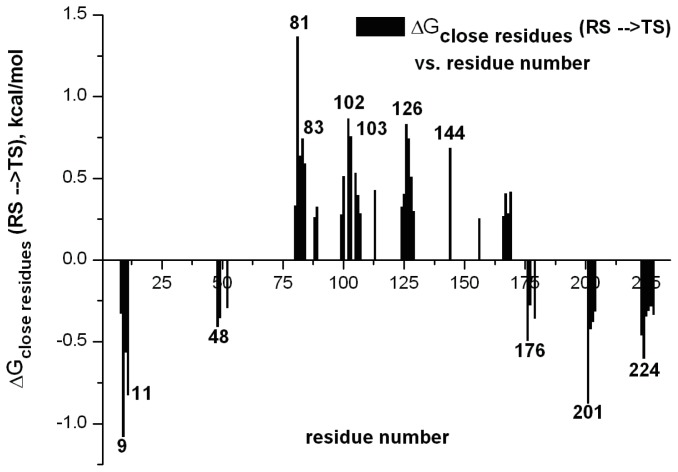
Enzymes are tremendously proficient catalysts, which can be used as extracellular catalysts for a whole host of processes, from chemical synthesis to the generation of novel biofuels. For them to be more amenable to the needs of biotechnology, however, it is often necessary to be able to manipulate their physico-chemical properties in an efficient and streamlined manner, and, ideally, to be able to train them to catalyze completely new reactions. Recent years have seen an explosion of interest in different approaches to achieve this, both in the laboratory, and in silico. There remains, however, a gap between current approaches to computational enzyme design, which have primarily focused on the early stages of the design process, and laboratory evolution, which is an extremely powerful tool for enzyme redesign, but will always be limited by the vastness of sequence space combined with the low frequency for desirable mutations. This review discusses different approaches towards computational enzyme design and demonstrates how combining newly developed screening approaches that can rapidly predict potential mutation "hotspots" with approaches that can quantitatively and reliably dissect the catalytic step can bridge the gap that currently exists between computational enzyme design and laboratory evolution studies.
酶是极其高效的催化剂,可作为细胞外催化剂用于从化学合成到新型生物燃料生成等一系列过程。然而,为了使其更符合生物技术的需求,通常需要能够以高效且简化的方式操纵它们的物理化学性质,理想情况下,还要能够训练它们催化全新的反应。近年来,无论是在实验室还是在计算机模拟中,人们对实现这一目标的不同方法兴趣激增。然而,当前的计算酶设计方法主要集中在设计过程的早期阶段,与实验室进化之间仍存在差距。实验室进化是酶重新设计的一种极其强大的工具,但总是会受到序列空间的巨大规模以及所需突变低频性的限制。本综述讨论了计算酶设计的不同方法,并展示了如何将能够快速预测潜在突变“热点”的新开发筛选方法与能够定量且可靠地剖析催化步骤的方法相结合,以弥合当前计算酶设计与实验室进化研究之间存在的差距。