De Simone Alfonso, Montalvao Rinaldo W, Vendruscolo Michele
J Chem Theory Comput. 2011 Dec 13;7(12):4189-4195. doi: 10.1021/ct200361b. Epub 2011 Oct 10.
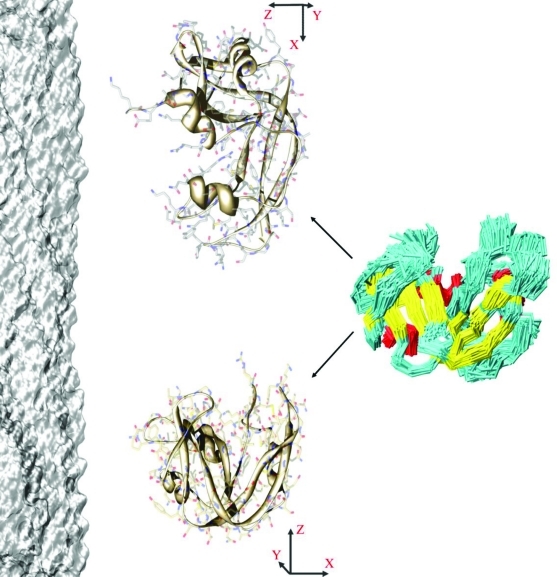
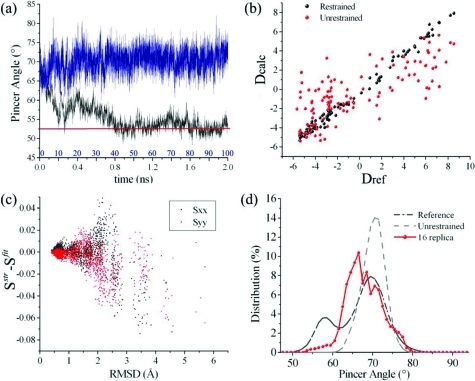
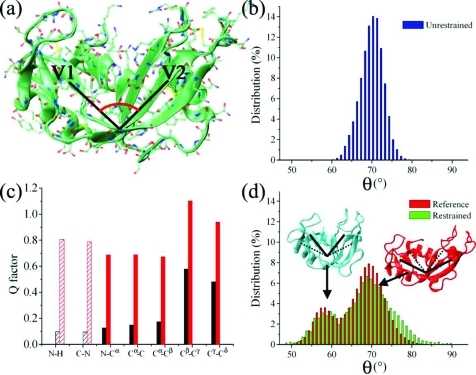
In order to carry out their functions, proteins often undergo significant conformational fluctuations that enable them to interact with their partners. The accurate characterization of these motions is key in order to understand the mechanisms by which macromolecular recognition events take place. Nuclear magnetic resonance spectroscopy offers a variety of powerful methods to achieve this result. We discuss a method of using residual dipolar couplings as replica-averaged restraints in molecular dynamics simulations to determine large amplitude motions of proteins, including those involved in the conformational equilibria that are established through interconversions between different states. By applying this method to ribonuclease A, we show that it enables one to characterize the ample fluctuations in interdomain orientations expected to play an important functional role.
为了执行其功能,蛋白质常常经历显著的构象波动,使其能够与它们的伙伴相互作用。准确表征这些运动是理解大分子识别事件发生机制的关键。核磁共振光谱提供了多种强大的方法来实现这一结果。我们讨论了一种在分子动力学模拟中使用残余偶极耦合作为复制平均约束来确定蛋白质大幅度运动的方法,包括那些通过不同状态之间的相互转换建立的构象平衡中涉及的运动。通过将这种方法应用于核糖核酸酶A,我们表明它能够表征预期发挥重要功能作用的结构域间取向的充分波动。