Center for Microbial Genetics and Genomics, Northern Arizona University, Flagstaff, Arizona, USA.
PLoS Negl Trop Dis. 2011 Dec;5(12):e1381. doi: 10.1371/journal.pntd.0001381. Epub 2011 Dec 13.
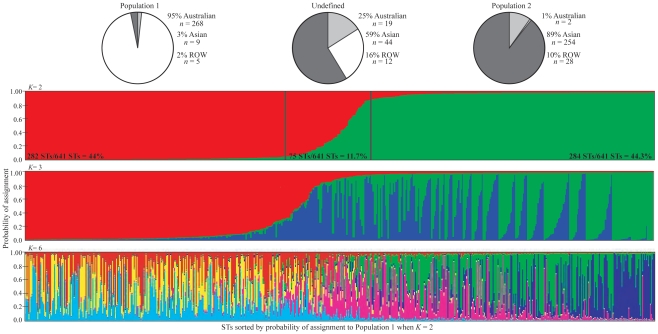
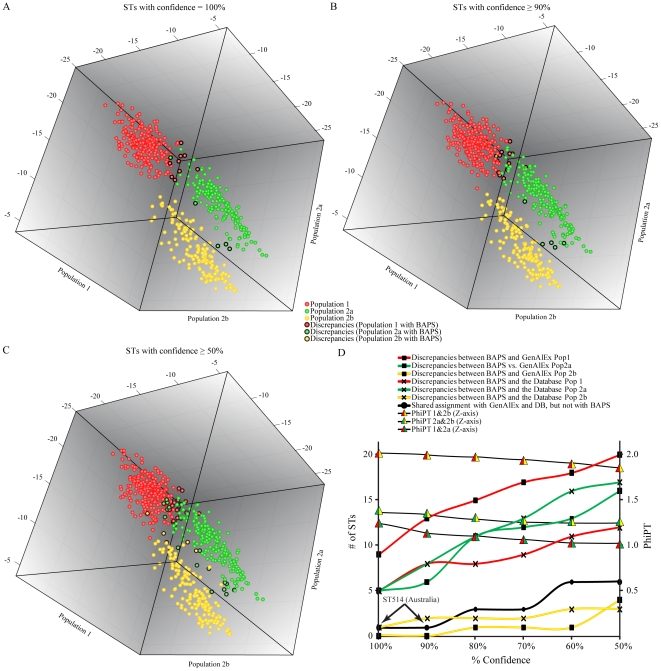
Rapid assignment of bacterial pathogens into predefined populations is an important first step for epidemiological tracking. For clonal species, a single allele can theoretically define a population. For non-clonal species such as Burkholderia pseudomallei, however, shared allelic states between distantly related isolates make it more difficult to identify population defining characteristics. Two distinct B. pseudomallei populations have been previously identified using multilocus sequence typing (MLST). These populations correlate with the major foci of endemicity (Australia and Southeast Asia). Here, we use multiple Bayesian approaches to evaluate the compositional robustness of these populations, and provide assignment results for MLST sequence types (STs). Our goal was to provide a reference for assigning STs to an established population without the need for further computational analyses. We also provide allele frequency results for each population to enable estimation of population assignment even when novel STs are discovered. The ability for humans and potentially contaminated goods to move rapidly across the globe complicates the task of identifying the source of an infection or outbreak. Population genetic dynamics of B. pseudomallei are particularly complicated relative to other bacterial pathogens, but the work here provides the ability for broad scale population assignment. As there is currently no independent empirical measure of successful population assignment, we provide comprehensive analytical details of our comparisons to enable the reader to evaluate the robustness of population designations and assignments as they pertain to individual research questions. Finer scale subdivision and verification of current population compositions will likely be possible with genotyping data that more comprehensively samples the genome. The approach used here may be valuable for other non-clonal pathogens that lack simple group-defining genetic characteristics and provides a rapid reference for epidemiologists wishing to track the origin of infection without the need to compile population data and learn population assignment algorithms.
快速将细菌病原体分配到预定义的群体中是进行流行病学追踪的重要第一步。对于克隆种,单个等位基因理论上可以定义一个群体。然而,对于非克隆种(如伯克霍尔德氏菌),由于远缘分离株之间存在共享等位基因状态,因此更难以确定种群定义特征。先前已经使用多位点序列分型(MLST)鉴定了两种不同的伯克霍尔德氏菌种群。这些种群与主要的流行(澳大利亚和东南亚)有关。在这里,我们使用多种贝叶斯方法来评估这些种群的组成稳健性,并为 MLST 序列型(ST)提供分配结果。我们的目标是提供一种参考,以便在无需进一步计算分析的情况下将 ST 分配给已建立的种群。我们还提供每个种群的等位基因频率结果,以便即使发现新的 ST,也能够估计种群分配。人类和潜在污染的货物能够迅速在全球范围内移动,这使得确定感染源或暴发源的任务变得复杂。与其他细菌病原体相比,伯克霍尔德氏菌的种群遗传动态特别复杂,但这里的工作为广泛的种群分配提供了能力。由于目前没有成功种群分配的独立经验衡量标准,因此我们提供了对我们比较的全面分析细节,以使读者能够评估种群指定和分配的稳健性,因为它们与个别研究问题有关。随着更全面地采样基因组的基因分型数据,更精细的细分和对当前种群组成的验证可能成为可能。这里使用的方法可能对其他缺乏简单群体定义遗传特征的非克隆病原体有价值,并为希望在无需编译群体数据和学习群体分配算法的情况下跟踪感染源的流行病学家提供快速参考。