Univ Paris-Sud, Institut de Génétique et Microbiologie, UMR 8621, Orsay, France.
PLoS One. 2011;6(12):e29190. doi: 10.1371/journal.pone.0029190. Epub 2011 Dec 29.
Investigation of the genetic diversity of Mycobacterium tuberculosis in China has shown that Beijing genotype strains play a dominant role in the tuberculosis (TB) epidemic. In order to examine the strain diversity in the whole country, and to study the evolutionary development of Beijing strains, we sought to genotype a large collection of isolates using different methods.
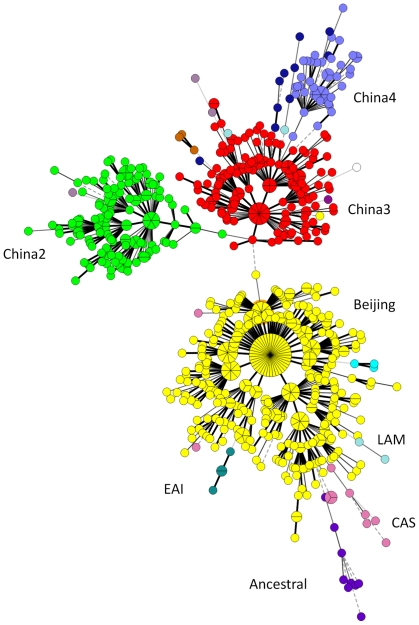
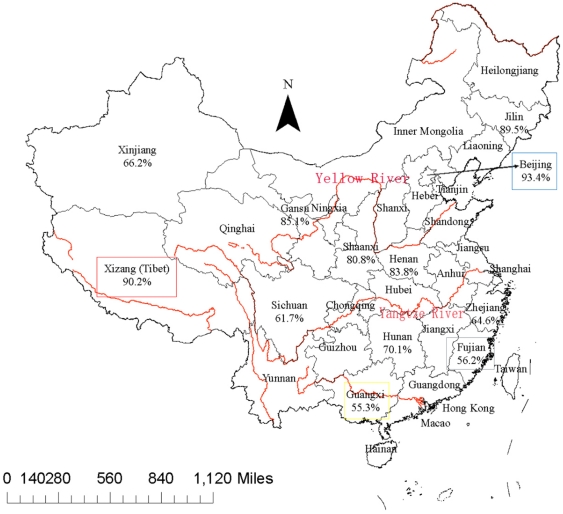
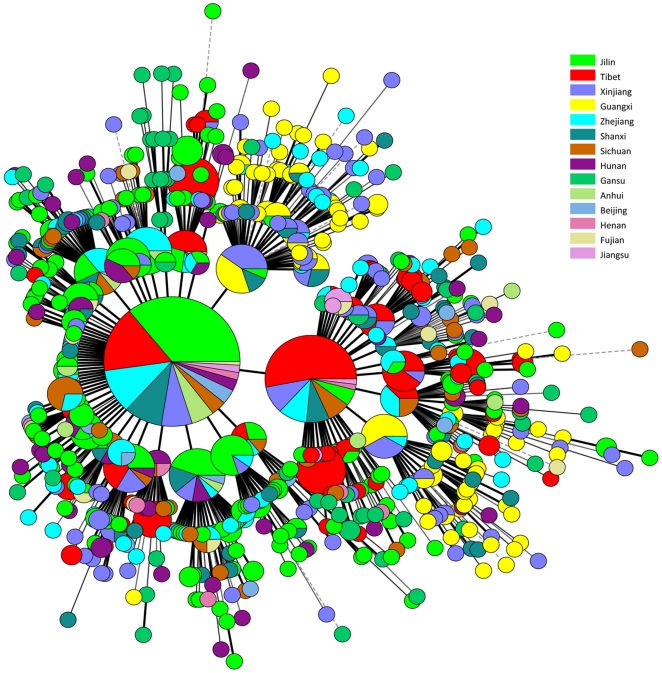
METHODOLOGY/PRINCIPAL FINDINGS: We applied a 15-loci VNTR typing analysis on 1,586 isolates from the Beijing municipality and 12 Chinese provinces or autonomous regions. The data was compared to that of 900 isolates from various other worldwide geographic regions outside of China. A total of 1,162/1,586 (73.2%) of the isolates, distributed into 472 VNTR types, were found to belong to the Beijing genotype family and this represented 56 to 94% of the isolates in each of the localizations. VNTR typing revealed that the majority of the non-Beijing isolates fall into two genotype families, which represented 17% of the total number of isolates, and seem largely restricted to China. A small number of East African Indian genotype strains was also observed in this collection. Ancient Beijing strains with an intact region of difference (RD) 181, as well as strains presumably resembling ancestors of the whole Beijing genotype family, were mainly found in the Guangxi autonomous region.
CONCLUSIONS/SIGNIFICANCE: This is the largest M. tuberculosis VNTR-based genotyping study performed in China to date. The high percentage of Beijing isolates in the whole country and the presence in the South of strains representing early branching points may be an indication that the Beijing lineage originated from China, probably in the Guangxi region. Two modern lineages are shown here to represent the majority of non-Beijing Chinese isolates. The observed geographic distribution of the different lineages within China suggests that natural frontiers are major factors in their diffusion.
对中国结核分枝杆菌遗传多样性的研究表明,北京基因型菌株在中国结核(TB)流行中占主导地位。为了全面考察全国的菌株多样性,并研究北京株的进化发展,我们试图使用不同方法对大量分离株进行基因分型。
方法/主要发现:我们对来自北京直辖市和中国 12 个省或自治区的 1586 株分离株进行了 15 个基因座 VNTR 分型分析。将数据与来自中国以外的 900 株来自世界各地不同地理区域的分离株进行比较。总共发现 1586 株分离株中的 1162 株(73.2%)属于北京基因型家族,其中包括来自各个地方的 56%至 94%的分离株。VNTR 分型显示,大多数非北京分离株属于两个基因型家族,占分离株总数的 17%,且似乎主要局限于中国。在本研究中还观察到少量东非印度基因型菌株。具有完整 RD181 区的古老北京株,以及可能类似于整个北京基因型家族祖先的菌株,主要发现于广西壮族自治区。
结论/意义:这是迄今为止在中国进行的最大规模的结核分枝杆菌基于 VNTR 的基因分型研究。全国范围内北京分离株的高比例,以及在南方存在代表早期分支点的菌株,可能表明北京谱系起源于中国,可能起源于广西地区。本研究表明,两种现代谱系代表了大多数非北京中国分离株。中国不同谱系的观察到的地理分布表明,自然边界是其传播的主要因素。