Department of Computing, Imperial College London , London, United Kingdom.
J Proteome Res. 2012 Mar 2;11(3):1571-81. doi: 10.1021/pr200698c. Epub 2012 Feb 9.
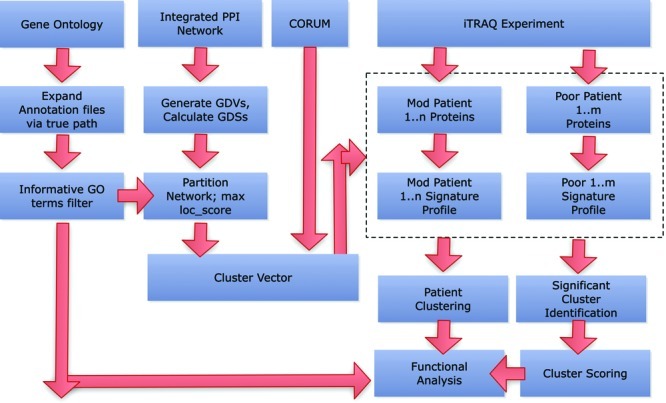
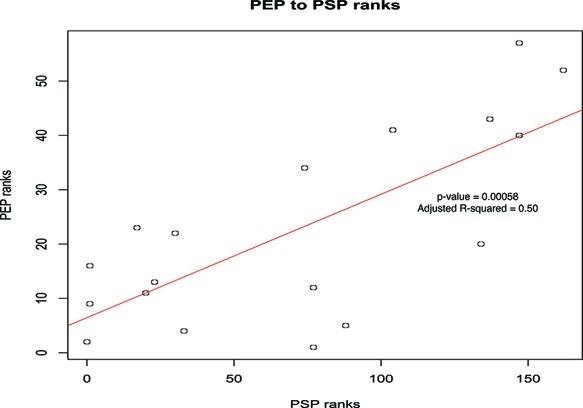
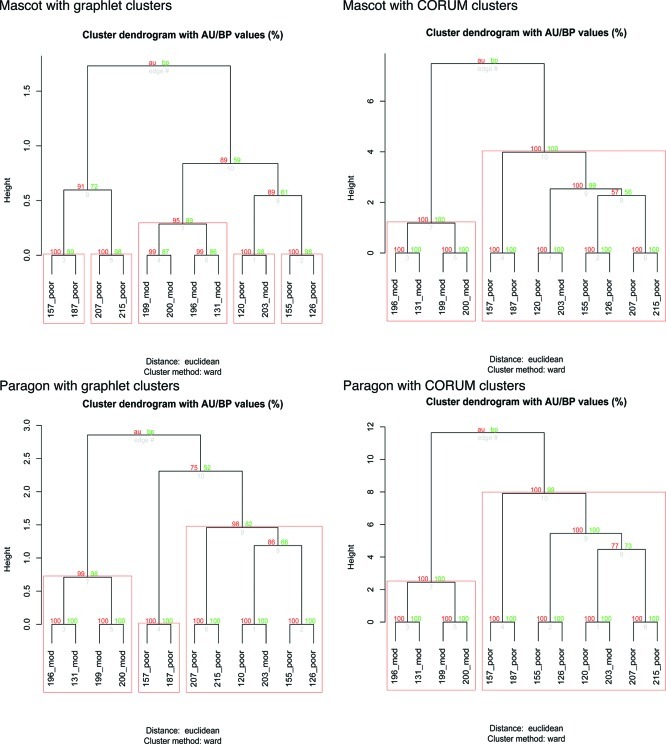
Traditional proteomics analysis is plagued by the use of arbitrary thresholds resulting in large loss of information. We propose here a novel method in proteomics that utilizes all detected proteins. We demonstrate its efficacy in a proteomics screen of 5 and 7 liver cancer patients in the moderate and late stage, respectively. Utilizing biological complexes as a cluster vector, and augmenting it with submodules obtained from partitioning an integrated and cleaned protein-protein interaction network, we calculate a Proteomics Signature Profile (PSP) for each patient based on the hit rates of their reported proteins, in the absence of fold change thresholds, against the cluster vector. Using this, we demonstrated that moderate- and late-stage patients segregate with high confidence. We also discovered a moderate-stage patient who displayed a proteomics profile similar to other poor-stage patients. We identified significant clusters using a modified version of the SNet approach. Comparing our results against the Proteomics Expansion Pipeline (PEP) on which the same patient data was analyzed, we found good correlation. Building on this finding, we report significantly more clusters (176 clusters here compared to 70 in PEP), demonstrating the sensitivity of this approach. Gene Ontology (GO) terms analysis also reveals that the significant clusters are functionally congruent with the liver cancer phenotype. PSP is a powerful and sensitive method for analyzing proteomics profiles even when sample sizes are small. It does not rely on the ratio scores but, rather, whether a protein is detected or not. Although consistency of individual proteins between patients is low, we found the reported proteins tend to hit clusters in a meaningful and informative manner. By extracting this information in the form of a Proteomics Signature Profile, we confirm that this information is conserved and can be used for (1) clustering of patient samples, (2) identification of significant clusters based on real biological complexes, and (3) overcoming consistency and coverage issues prevalent in proteomics data sets.
传统的蛋白质组学分析受到任意阈值的限制,导致大量信息丢失。我们在这里提出了一种新的蛋白质组学方法,该方法利用了所有检测到的蛋白质。我们在对分别处于中晚期的 5 名和 7 名肝癌患者的蛋白质组学筛选中证明了其功效。我们利用生物复合物作为聚类向量,并利用从集成和清理的蛋白质-蛋白质相互作用网络中划分得到的子模块对其进行扩充,根据报道的蛋白质的命中率(不使用倍数变化阈值)针对聚类向量为每位患者计算蛋白质组学特征图谱 (PSP)。使用这种方法,我们证明了中晚期患者能够很好地分离。我们还发现了一名中晚期患者,其蛋白质组学图谱与其他晚期患者相似。我们使用 SNet 方法的修改版本发现了显著的聚类。将我们的结果与基于相同患者数据进行分析的 Proteomics Expansion Pipeline (PEP) 进行比较,我们发现相关性很好。在此基础上,我们报告了更多的聚类(这里有 176 个聚类,而 PEP 中有 70 个聚类),证明了这种方法的敏感性。GO 术语分析还表明,显著聚类与肝癌表型在功能上是一致的。PSP 是一种强大而敏感的方法,即使样本量较小,也可以用于分析蛋白质组学图谱。它不依赖于比值分数,而是依赖于是否检测到蛋白质。尽管患者之间单个蛋白质的一致性较低,但我们发现报道的蛋白质倾向于以有意义和信息丰富的方式命中聚类。通过以蛋白质组学特征图谱的形式提取这些信息,我们确认该信息是保守的,可以用于 (1) 患者样本的聚类,(2) 根据真实的生物复合物识别显著聚类,以及 (3) 克服蛋白质组学数据集中普遍存在的一致性和覆盖范围问题。