Computational Biology Research Center (CBRC), National Institute of Advanced Industrial Science and Technology (AIST), 2-4-7 Aomi, Koto Ward, Tokyo 135-0064, Japan.
BMC Bioinformatics. 2012 Jan 16;13:11. doi: 10.1186/1471-2105-13-11.
Evolutionary relations of similar segments shared by different protein folds remain controversial, even though many examples of such segments have been found. To date, several methods such as those based on the results of structure comparisons, sequence-based classifications, and sequence-based profile-profile comparisons have been applied to identify such protein segments that possess local similarities in both sequence and structure across protein folds. However, to capture more precise sequence-structure relations, no method reported to date combines structure-based profiles, and sequence-based profiles based on evolutionary information. The former are generally regarded as representing the amino acid preferences at each position of a specific conformation of protein segment. They might reflect the nature of ancient short peptide ancestors, using the results of structural classifications of protein segments.
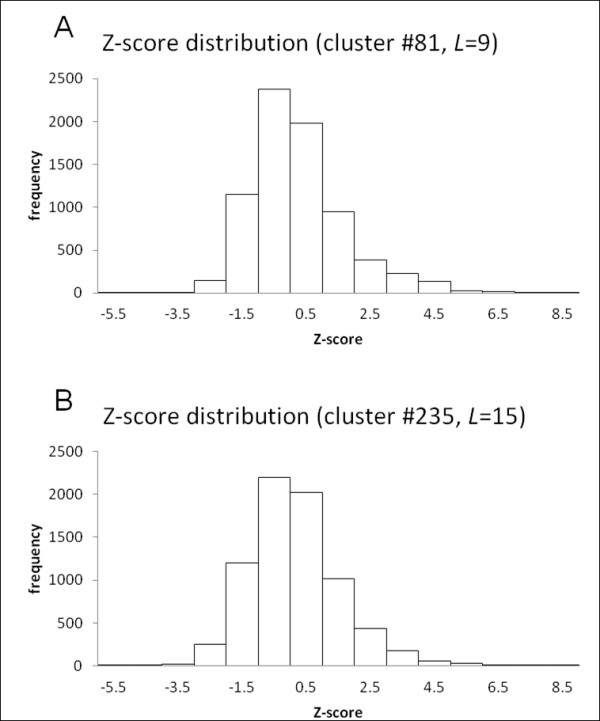
This report describes the development and use of "Cross Profile Analysis" to compare sequence-based profiles and structure-based profiles based on amino acid occurrences at each position within a protein segment cluster. Using systematic cross profile analysis, we found structural clusters of 9-residue and 15-residue segments showing remarkably strong correlation with particular sequence profiles. These correlations reflect structural similarities among constituent segments of both sequence-based and structure-based profiles. We also report previously undetectable sequence-structure patterns that transcend protein family and fold boundaries, and present results of the conformational analysis of the deduced peptide of a segment cluster. These results suggest the existence of ancient short-peptide ancestors.
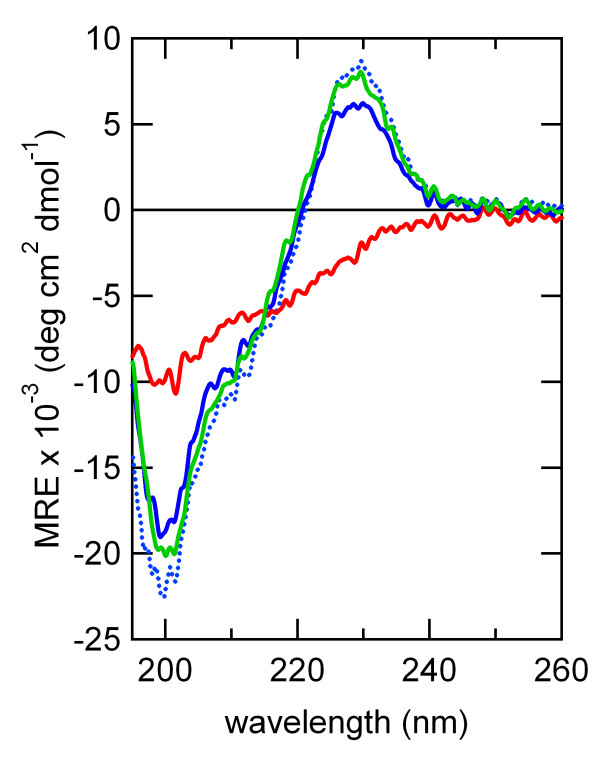
Cross profile analysis reveals the polyphyletic and convergent evolution of β-hairpin-like structures, which were verified both experimentally and computationally. The results presented here give us new insights into the evolution of short protein segments.
尽管已经发现了许多具有相似结构的蛋白质片段,但不同蛋白质折叠之间共享的相似片段的进化关系仍然存在争议。迄今为止,已经应用了几种方法,例如基于结构比较结果的方法、基于序列的分类方法和基于序列的轮廓-轮廓比较方法,来识别具有局部相似性的蛋白质片段,这些片段在蛋白质折叠中具有序列和结构上的相似性。然而,为了捕捉更精确的序列-结构关系,目前尚无方法将基于结构的轮廓和基于进化信息的基于序列的轮廓结合起来。前者通常被认为代表蛋白质片段特定构象中每个位置的氨基酸偏好。它们可能反映了古老短肽祖先的性质,使用蛋白质片段结构分类的结果。
本报告描述了“交叉轮廓分析”的开发和使用,以比较基于序列的轮廓和基于结构的轮廓,这些轮廓基于蛋白质片段簇中每个位置的氨基酸出现情况。使用系统的交叉轮廓分析,我们发现了具有 9 个残基和 15 个残基的结构簇与特定序列轮廓显示出非常强的相关性。这些相关性反映了基于序列和基于结构的轮廓中组成片段的结构相似性。我们还报告了以前无法检测到的超越蛋白质家族和折叠边界的序列-结构模式,并呈现了推断出的片段簇的肽的构象分析结果。这些结果表明存在古老的短肽祖先。
交叉轮廓分析揭示了β发夹样结构的多系和趋同进化,这在实验和计算上都得到了验证。这里呈现的结果使我们对短蛋白质片段的进化有了新的认识。