Department of Genome Sciences, University of Washington , 815 Mercer Street, Seattle, Washington 98109, United States.
J Proteome Res. 2012 Mar 2;11(3):1621-32. doi: 10.1021/pr2008175. Epub 2012 Feb 21.
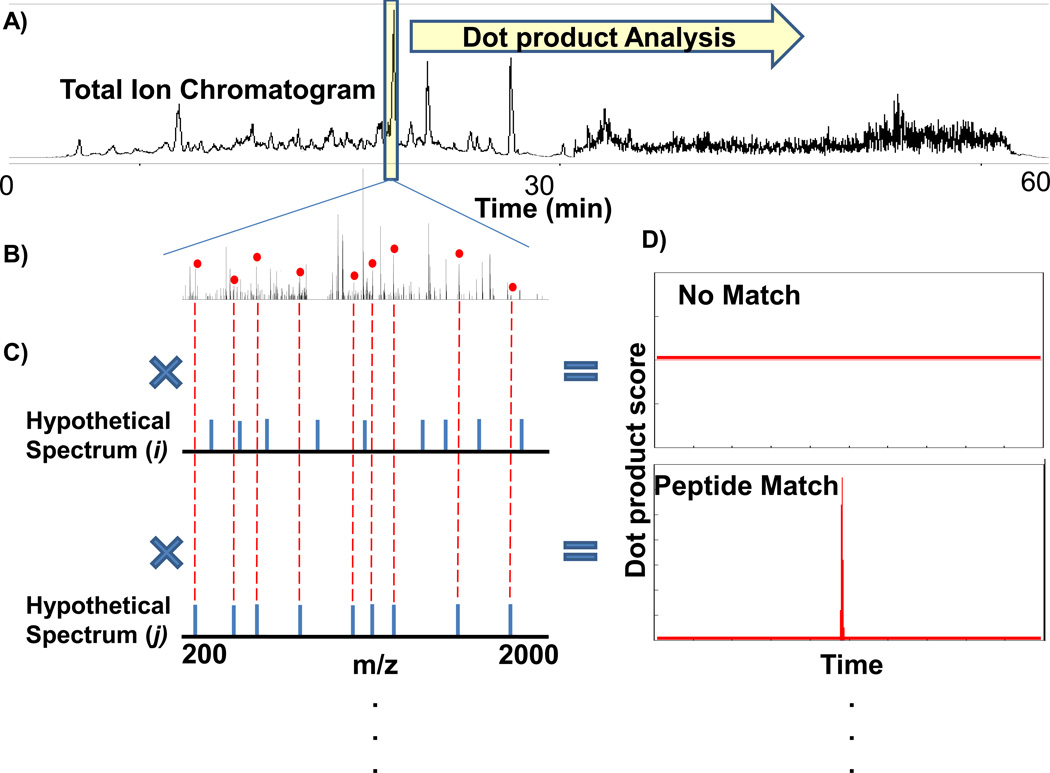
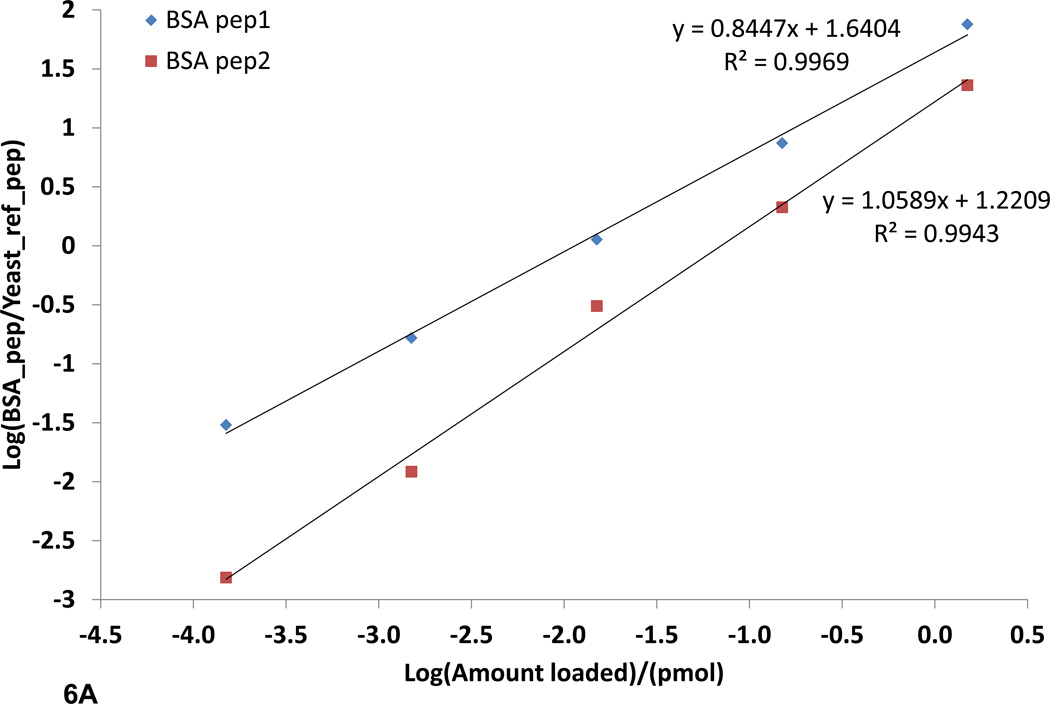
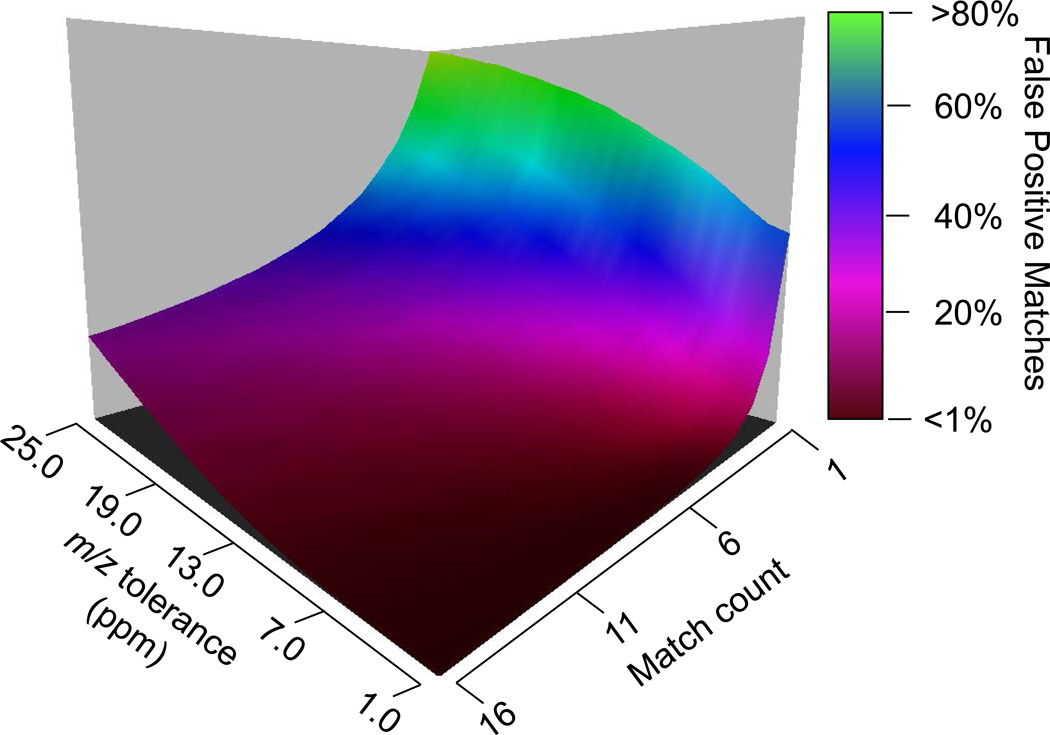
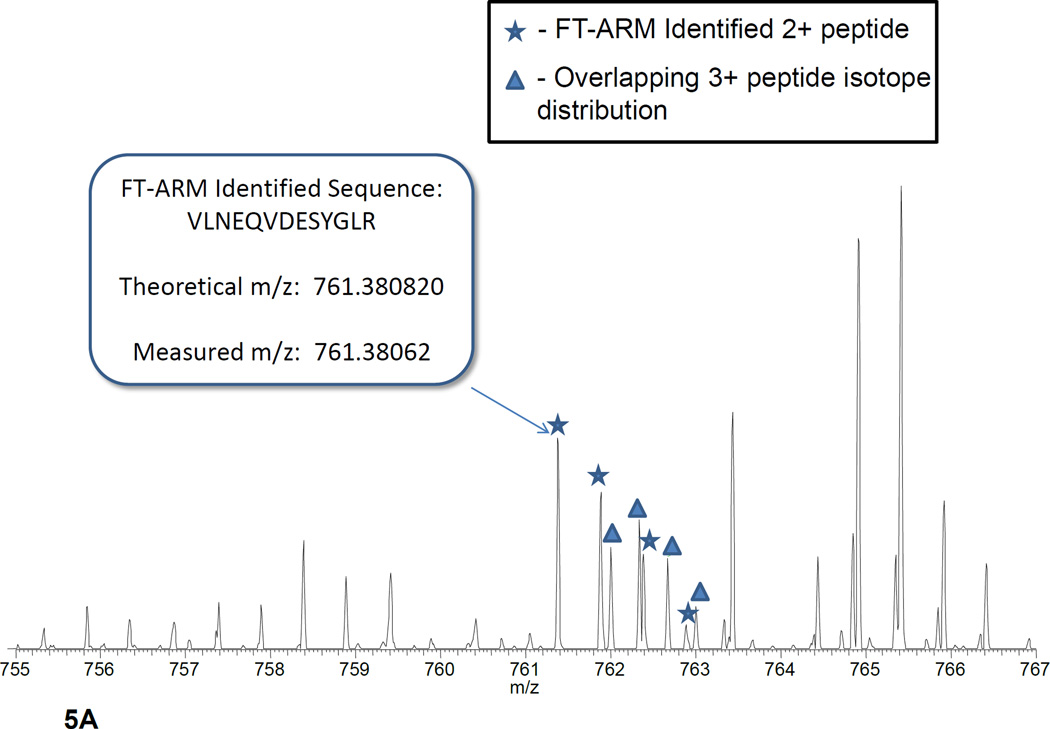
Fourier transform-all reaction monitoring (FT-ARM) is a novel approach for the identification and quantification of peptides that relies upon the selectivity of high mass accuracy data and the specificity of peptide fragmentation patterns. An FT-ARM experiment involves continuous, data-independent, high mass accuracy MS/MS acquisition spanning a defined m/z range. Custom software was developed to search peptides against the multiplexed fragmentation spectra by comparing theoretical or empirical fragment ions against every fragmentation spectrum across the entire acquisition. A dot product score is calculated against each spectrum to generate a score chromatogram used for both identification and quantification. Chromatographic elution profile characteristics are not used to cluster precursor peptide signals to their respective fragment ions. FT-ARM identifications are demonstrated to be complementary to conventional data-dependent shotgun analysis, especially in cases where the data-dependent method fails because of fragmenting multiple overlapping precursors. The sensitivity, robustness, and specificity of FT-ARM quantification are shown to be analogous to selected reaction monitoring-based peptide quantification with the added benefit of minimal assay development. Thus, FT-ARM is demonstrated to be a novel and complementary data acquisition, identification, and quantification method for the large scale analysis of peptides.
傅里叶变换全反应监测(FT-ARM)是一种用于鉴定和定量肽的新方法,它依赖于高质量精度数据的选择性和肽碎片模式的特异性。FT-ARM 实验涉及连续的、独立的数据、高质量精度的 MS/MS 采集,跨越定义的 m/z 范围。开发了自定义软件,通过将理论或经验碎片离子与整个采集过程中的每个碎片光谱进行比较,搜索肽对多路碎片光谱的匹配。对每个光谱计算一个点积分数,以生成用于鉴定和定量的得分色谱图。不使用色谱洗脱轮廓特征将前体肽信号聚类到各自的碎片离子。FT-ARM 的鉴定被证明与传统的基于数据依赖性的 shotgun 分析是互补的,特别是在数据依赖性方法由于碎裂多个重叠的前体而失败的情况下。FT-ARM 定量的灵敏度、稳健性和特异性与基于选择反应监测的肽定量类似,其优点是最小化了测定的开发。因此,FT-ARM 被证明是一种用于大规模分析肽的新的和互补的数据采集、鉴定和定量方法。