National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, United States of America.
PLoS One. 2012;7(1):e28896. doi: 10.1371/journal.pone.0028896. Epub 2012 Jan 31.
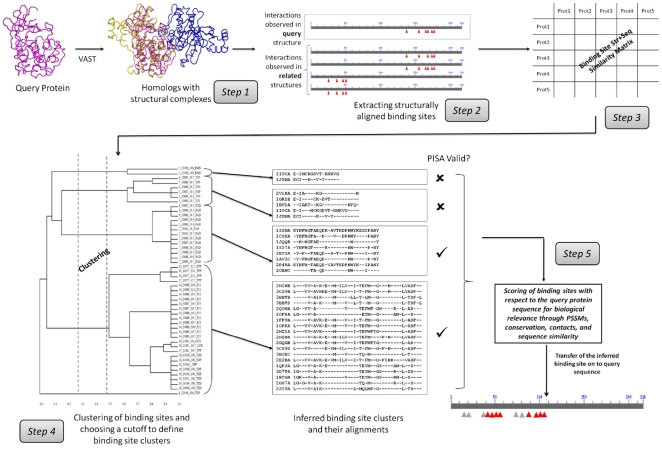
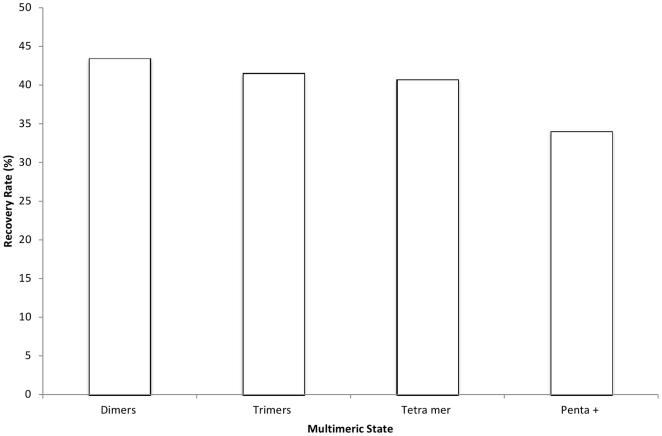
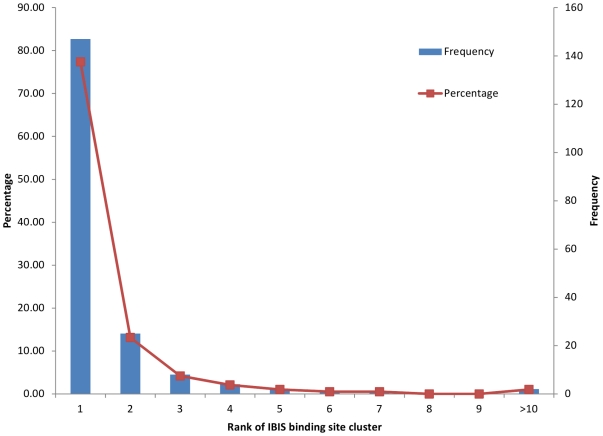
The coverage and reliability of protein-protein interactions determined by high-throughput experiments still needs to be improved, especially for higher organisms, therefore the question persists, how interactions can be verified and predicted by computational approaches using available data on protein structural complexes. Recently we developed an approach called IBIS (Inferred Biomolecular Interaction Server) to predict and annotate protein-protein binding sites and interaction partners, which is based on the assumption that the structural location and sequence patterns of protein-protein binding sites are conserved between close homologs. In this study first we confirmed high accuracy of our method and found that its accuracy depends critically on the usage of all available data on structures of homologous complexes, compared to the approaches where only a non-redundant set of complexes is employed. Second we showed that there exists a trade-off between specificity and sensitivity if we employ in the prediction only evolutionarily conserved binding site clusters or clusters supported by only one observation (singletons). Finally we addressed the question of identifying the biologically relevant interactions using the homology inference approach and demonstrated that a large majority of crystal packing interactions can be correctly identified and filtered by our algorithm. At the same time, about half of biological interfaces that are not present in the protein crystallographic asymmetric unit can be reconstructed by IBIS from homologous complexes without the prior knowledge of crystal parameters of the query protein.
高通量实验确定的蛋白质-蛋白质相互作用的覆盖范围和可靠性仍有待提高,特别是对于高等生物,因此仍然存在一个问题,即如何使用可用的蛋白质结构复合物数据通过计算方法来验证和预测相互作用。最近,我们开发了一种称为 IBIS(推断生物分子相互作用服务器)的方法来预测和注释蛋白质-蛋白质结合位点和相互作用伙伴,该方法基于这样一种假设,即蛋白质-蛋白质结合位点的结构位置和序列模式在密切同源物之间是保守的。在这项研究中,我们首先证实了我们方法的高精度,并且发现与仅使用非冗余复合物集的方法相比,其准确性严重依赖于对同源复合物结构的所有可用数据的使用。其次,如果我们仅使用进化保守的结合位点簇或仅由一个观察结果(单峰)支持的簇来进行预测,那么特异性和灵敏度之间存在权衡。最后,我们使用同源推断方法解决了识别生物学相关相互作用的问题,并证明了我们的算法可以正确识别和过滤大量晶体包装相互作用。同时,通过 IBIS 可以从同源复合物中重建约一半未出现在蛋白质晶体学不对称单元中的生物界面,而无需查询蛋白质的晶体参数的先验知识。