Department of Microbiology and Molecular Genetics and the Center for Microbial Pathogenesis, Michigan State University, East Lansing, MI, USA.
BMC Microbiol. 2012 Feb 7;12:20. doi: 10.1186/1471-2180-12-20.
Porcine tonsils are the colonization site for many pathogenic as well as commensal microorganisms and are the primary lymphoid tissue encountered by organisms entering through the mouth or nares. The goal of this study was to provide an in-depth characterization of the composition and structure of the tonsillar microbial communities and to define the core microbiome in the tonsils of healthy pigs, using high throughput bar-coded 454-FLX pyrosequencing.
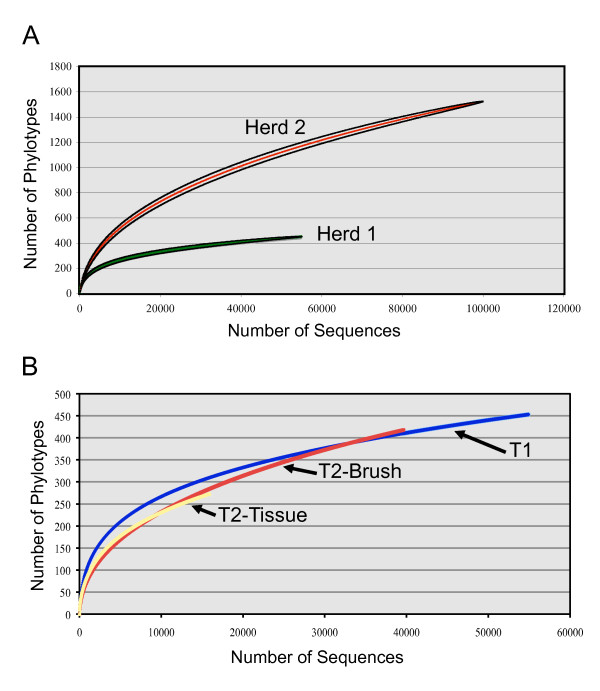
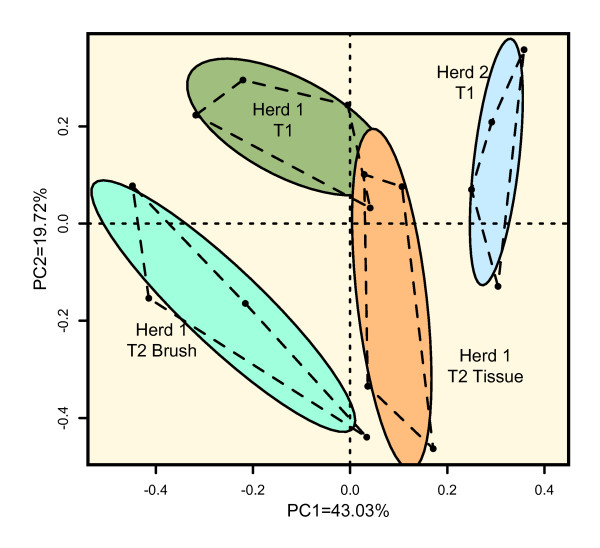
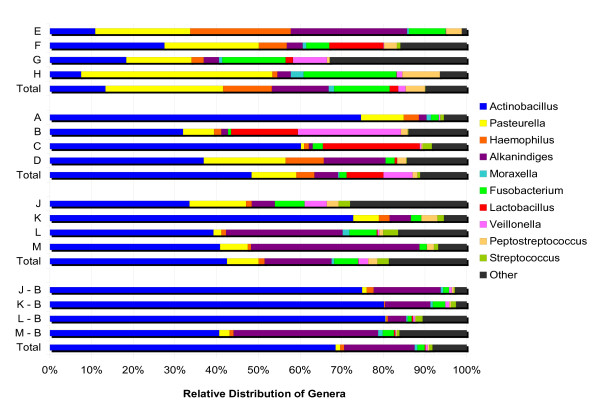
Whole tonsils were collected at necropsy from 12 16-week-old finisher pigs from two healthy herds. Tonsil brushes were also used to collect samples from four of these animals. Bacterial DNA was isolated from each sample, amplified by PCR with universal primers specific for the bacterial 16S rRNA genes, and the PCR products sequenced using pyrosequencing. An average of 13,000 sequences were generated from each sample. Microbial community members were identified by sequence comparison to known bacterial 16S rRNA gene sequences.The microbiomes of these healthy herds showed very strong similarities in the major components as well as distinct differences in minor components. Pasteurellaceae dominated the tonsillar microbiome in all animals, comprising ~60% of the total, although the relative proportions of the genera Actinobacillus, Haemophilus, and Pasteurella varied between the herds. Also found in all animals were the genera Alkanindiges, Peptostreptococcus, Veillonella, Streptococcus and Fusobacterium, as well as Enterobacteriaceae and Neisseriaceae. Treponema and Chlamydia were unique to Herd 1, while Arcanobacterium was unique to Herd 2.Tonsil brushes yielded similar results to tissue specimens, although Enterobacteriaceae and obligate anaerobes were more frequently found in tissue than in brush samples, and Chlamydia, an obligately intracellular organism, was not found in brush specimens.
We have extended and supported our previous studies with 16S clone libraries, using 16S rRNA gene pyrosequencing to describe the microbial communities in tonsils of healthy pigs. We have defined a core microbiome, dominated by Pasteurellaceae, in tonsil specimens, and have also demonstrated the presence of unique minor components of the tonsillar microbiome present in each herd. We have validated the use of non-invasive tonsil brushes, in comparison to tonsil tissue, which will facilitate future studies.
猪扁桃体是许多病原微生物和共生微生物的定植部位,也是通过口腔或鼻腔进入的生物体首先遇到的主要淋巴组织。本研究的目的是通过高通量 bar-coded 454-FLX 焦磷酸测序,深入描述扁桃体微生物群落的组成和结构,并确定健康猪扁桃体的核心微生物组。
从两个健康猪群的 12 头 16 周龄育肥猪的尸检中采集整个扁桃体。还使用扁桃体刷从其中 4 只动物采集样本。从每个样本中提取细菌 DNA,用针对细菌 16S rRNA 基因的通用引物进行 PCR 扩增,并使用焦磷酸测序对 PCR 产物进行测序。从每个样本中平均生成 13000 个序列。通过与已知细菌 16S rRNA 基因序列的序列比较来鉴定微生物群落成员。这些健康猪群的微生物组在主要成分上非常相似,在次要成分上则存在明显差异。巴氏杆菌科在所有动物的扁桃体微生物组中占主导地位,占总数的 60%左右,尽管两个猪群中放线杆菌属、嗜血杆菌属和巴氏杆菌属的相对比例有所不同。在所有动物中还发现了 Alkanindiges、Peptostreptococcus、Veillonella、Streptococcus 和 Fusobacterium 以及肠杆菌科和奈瑟菌科。螺旋体和衣原体是 1 号群特有的,而 Arcanobacterium 是 2 号群特有的。扁桃体刷的结果与组织标本相似,尽管组织样本中比刷子样本更常发现肠杆菌科和严格厌氧菌,而且严格的细胞内生物衣原体在刷子样本中未发现。
我们使用 16S rRNA 基因焦磷酸测序扩展并支持了我们之前使用 16S 克隆文库的研究,描述了健康猪扁桃体的微生物群落。我们在扁桃体标本中定义了一个以巴氏杆菌科为主导的核心微生物组,并且还证明了每个猪群存在独特的扁桃体微生物组次要成分。我们验证了与扁桃体组织相比,非侵入性的扁桃体刷的使用,这将方便未来的研究。