Department of Biostatistics, Indiana University, Indianapolis, IN 46202, USA.
BMC Bioinformatics. 2012 Feb 8;13:27. doi: 10.1186/1471-2105-13-27.
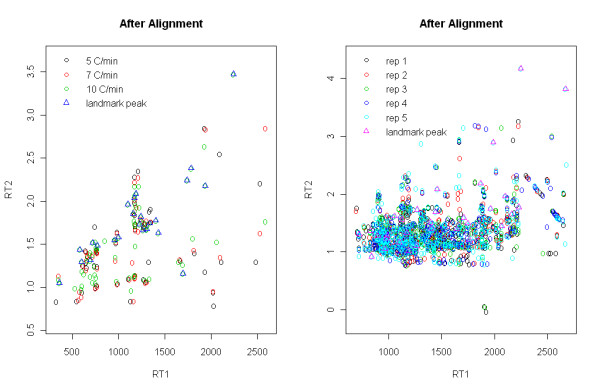
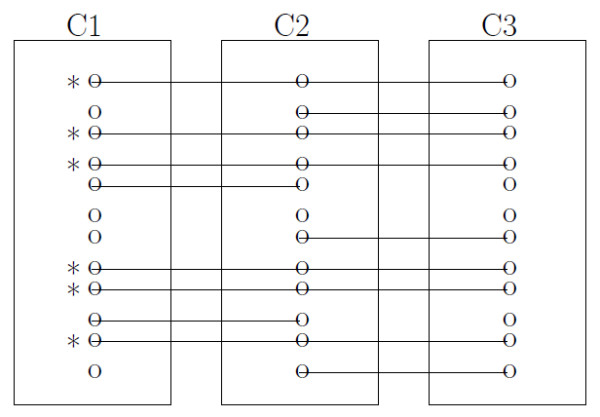
Comprehensive two-dimensional gas chromatography time-of-flight mass spectrometry (GCxGC/TOF-MS) has been used for metabolite profiling in metabolomics. However, there is still much experimental variation to be controlled including both within-experiment and between-experiment variation. For efficient analysis, an ideal peak alignment method to deal with such variations is in great need.
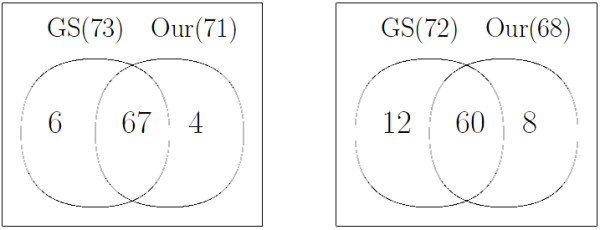
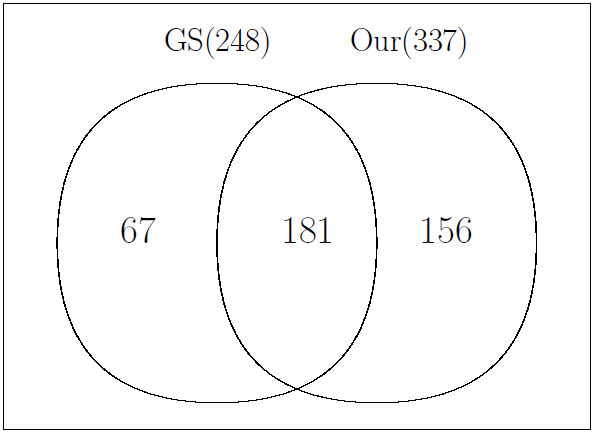
Using experimental data of a mixture of metabolite standards, we demonstrated that our method has better performance than other existing method which is not model-based. We then applied our method to the data generated from the plasma of a rat, which also demonstrates good performance of our model.
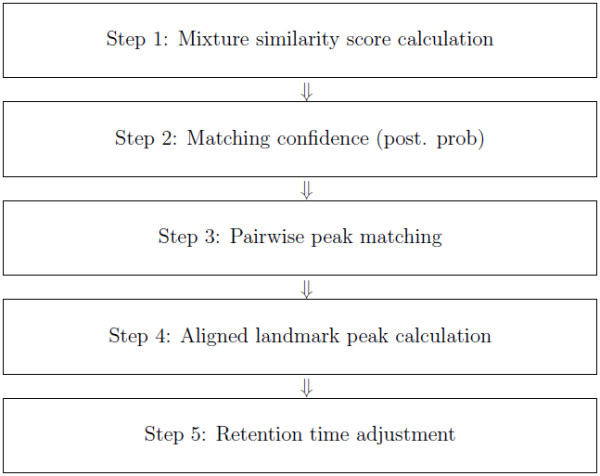
We developed a model-based peak alignment method to process both homogeneous and heterogeneous experimental data. The unique feature of our method is the only model-based peak alignment method coupled with metabolite identification in an unified framework. Through the comparison with other existing method, we demonstrated that our method has better performance. Data are available at http://stage.louisville.edu/faculty/x0zhan17/software/software-development/mspa. The R source codes are available at http://www.biostat.iupui.edu/~ChangyuShen/CodesPeakAlignment.zip.
2136949528613691.
全面二维气相色谱飞行时间质谱(GCxGC/TOF-MS)已用于代谢组学中的代谢物分析。然而,仍有许多实验变化需要控制,包括实验内和实验间的变化。为了进行有效的分析,需要一种理想的峰对齐方法来处理这些变化。
使用代谢物标准混合物的实验数据,我们证明了我们的方法比其他非基于模型的现有方法具有更好的性能。然后,我们将我们的方法应用于大鼠血浆产生的数据,也证明了我们的模型具有良好的性能。
我们开发了一种基于模型的峰对齐方法来处理同质性和异质性实验数据。我们的方法的独特之处在于,它是唯一的基于模型的峰对齐方法,与代谢物鉴定结合在一个统一的框架中。通过与其他现有方法的比较,我们证明了我们的方法具有更好的性能。数据可在 http://stage.louisville.edu/faculty/x0zhan17/software/software-development/mspa 上获得。R 源代码可在 http://www.biostat.iupui.edu/~ChangyuShen/CodesPeakAlignment.zip 上获得。
2136949528613691。