DE Shaw Research, New York, New York, United States of America.
PLoS One. 2012;7(2):e32131. doi: 10.1371/journal.pone.0032131. Epub 2012 Feb 22.
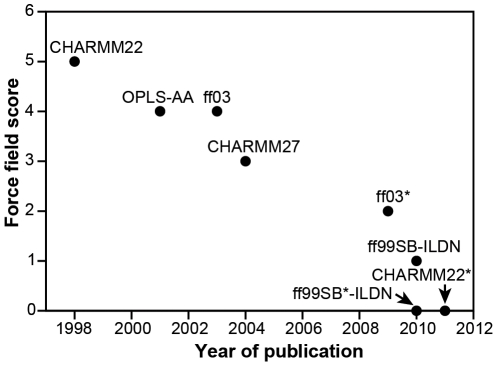
Molecular dynamics simulations provide a vehicle for capturing the structures, motions, and interactions of biological macromolecules in full atomic detail. The accuracy of such simulations, however, is critically dependent on the force field--the mathematical model used to approximate the atomic-level forces acting on the simulated molecular system. Here we present a systematic and extensive evaluation of eight different protein force fields based on comparisons of experimental data with molecular dynamics simulations that reach a previously inaccessible timescale. First, through extensive comparisons with experimental NMR data, we examined the force fields' abilities to describe the structure and fluctuations of folded proteins. Second, we quantified potential biases towards different secondary structure types by comparing experimental and simulation data for small peptides that preferentially populate either helical or sheet-like structures. Third, we tested the force fields' abilities to fold two small proteins--one α-helical, the other with β-sheet structure. The results suggest that force fields have improved over time, and that the most recent versions, while not perfect, provide an accurate description of many structural and dynamical properties of proteins.
分子动力学模拟为捕捉生物大分子的结构、运动和相互作用提供了一个工具,其可以达到全原子细节的程度。然而,这种模拟的准确性严重依赖于力场——即用于近似模拟分子系统中原子级力的数学模型。在这里,我们基于与分子动力学模拟的比较,对 8 种不同的蛋白质力场进行了系统而广泛的评估,这些模拟达到了以前无法达到的时间尺度。首先,通过与实验 NMR 数据的广泛比较,我们考察了力场描述折叠蛋白质结构和波动的能力。其次,我们通过比较优先形成螺旋或片状结构的小肽的实验和模拟数据,量化了对不同二级结构类型的潜在偏差。第三,我们测试了力场折叠两个小蛋白的能力——一个是α-螺旋,另一个是β-折叠结构。结果表明,力场随着时间的推移而不断改进,尽管最近的版本并不完美,但它们提供了对蛋白质许多结构和动力学性质的准确描述。