Information Engineering Department, University of Padova, Padova, Italy.
PLoS One. 2012;7(3):e32200. doi: 10.1371/journal.pone.0032200. Epub 2012 Mar 5.
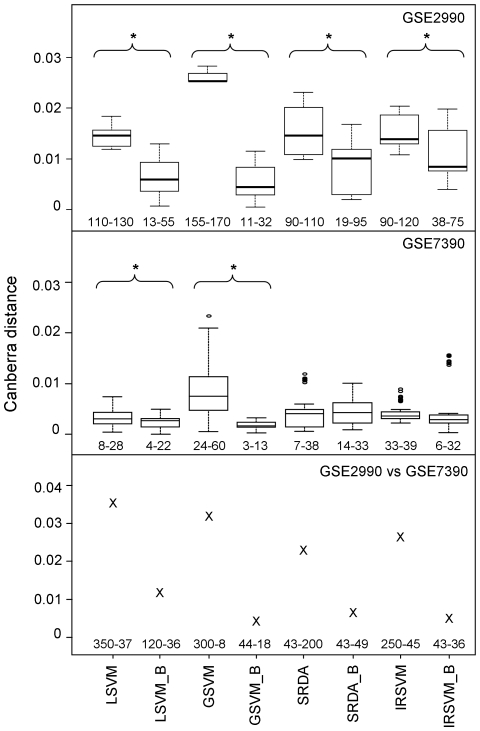
The identification of robust lists of molecular biomarkers related to a disease is a fundamental step for early diagnosis and treatment. However, methodologies for the discovery of biomarkers using microarray data often provide results with limited overlap. These differences are imputable to 1) dataset size (few subjects with respect to the number of features); 2) heterogeneity of the disease; 3) heterogeneity of experimental protocols and computational pipelines employed in the analysis. In this paper, we focus on the first two issues and assess, both on simulated (through an in silico regulation network model) and real clinical datasets, the consistency of candidate biomarkers provided by a number of different methods.
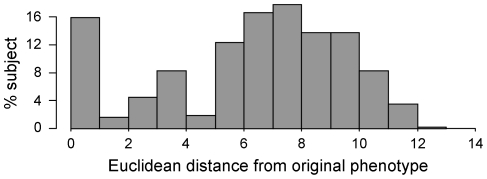
We extensively simulated the effect of heterogeneity characteristic of complex diseases on different sets of microarray data. Heterogeneity was reproduced by simulating both intrinsic variability of the population and the alteration of regulatory mechanisms. Population variability was simulated by modeling evolution of a pool of subjects; then, a subset of them underwent alterations in regulatory mechanisms so as to mimic the disease state.
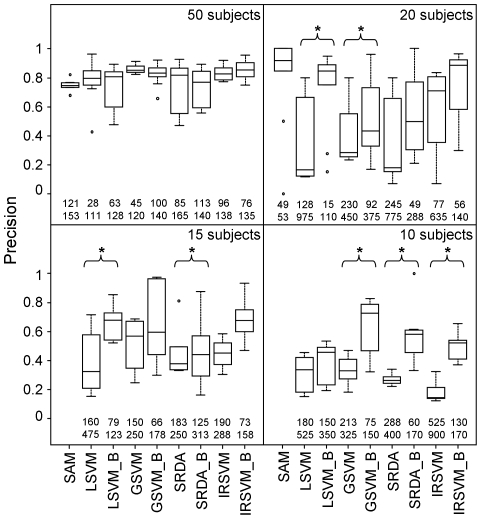
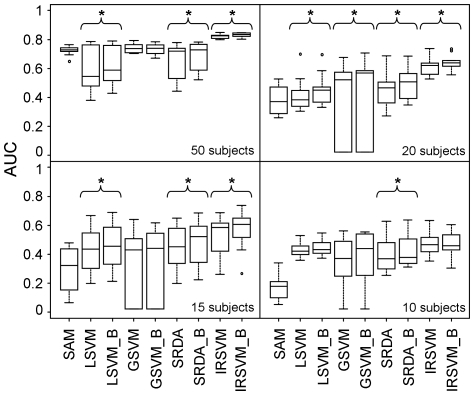
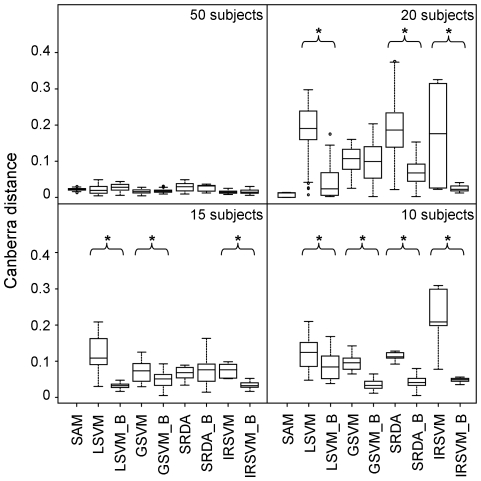
The simulated data allowed us to outline advantages and drawbacks of different methods across multiple studies and varying number of samples and to evaluate precision of feature selection on a benchmark with known biomarkers. Although comparable classification accuracy was reached by different methods, the use of external cross-validation loops is helpful in finding features with a higher degree of precision and stability. Application to real data confirmed these results.
鉴定与疾病相关的稳健分子生物标志物列表是早期诊断和治疗的基础步骤。然而,使用微阵列数据发现生物标志物的方法通常提供的结果重叠有限。这些差异可归因于 1)数据集大小(相对于特征数量,样本数量较少);2)疾病的异质性;3)分析中使用的实验方案和计算管道的异质性。在本文中,我们专注于前两个问题,并在模拟(通过计算机调控网络模型)和真实临床数据集上评估了许多不同方法提供的候选生物标志物的一致性。
我们广泛模拟了复杂疾病特有的异质性对不同微阵列数据集的影响。通过模拟群体的固有变异性和调控机制的改变来再现异质性。通过对一组主题的演变进行建模来模拟群体变异性;然后,其中一部分经历了调控机制的改变,以模拟疾病状态。
模拟数据使我们能够在多个研究中概述不同方法的优缺点,以及在不同数量的样本和评估具有已知生物标志物的基准特征选择精度方面的优势。尽管不同方法达到了可比的分类准确性,但使用外部交叉验证循环有助于找到具有更高精度和稳定性的特征。对真实数据的应用证实了这些结果。