The University of Queensland, Institute for Molecular Bioscience, 306 Carmody Road, St Lucia, Brisbane, Queensland 4072, Australia.
Genome Med. 2012 May 1;4(5):41. doi: 10.1186/gm340.
Altered networks of gene regulation underlie many complex conditions, including cancer. Inferring gene regulatory networks from high-throughput microarray expression data is a fundamental but challenging task in computational systems biology and its translation to genomic medicine. Although diverse computational and statistical approaches have been brought to bear on the gene regulatory network inference problem, their relative strengths and disadvantages remain poorly understood, largely because comparative analyses usually consider only small subsets of methods, use only synthetic data, and/or fail to adopt a common measure of inference quality.
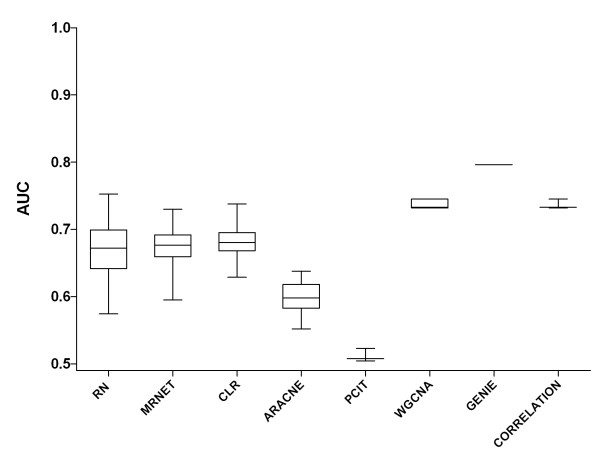
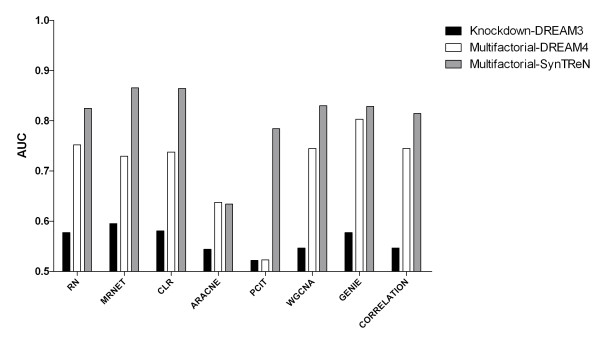
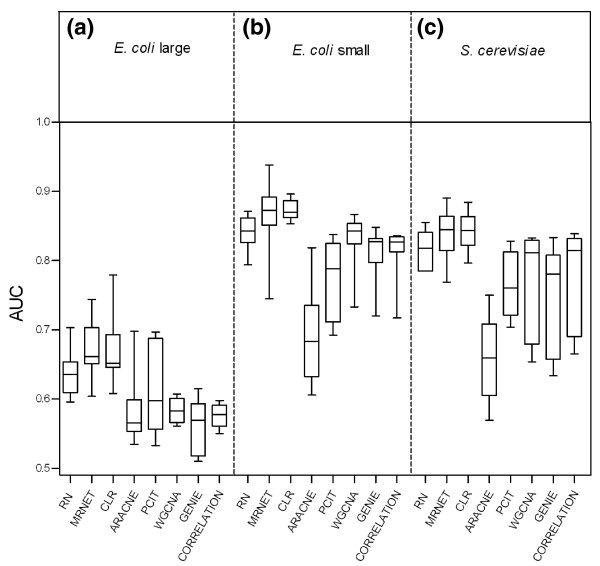
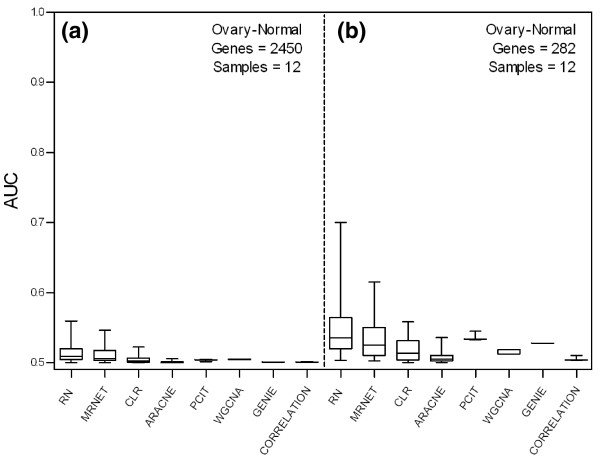
We report a comprehensive comparative evaluation of nine state-of-the art gene regulatory network inference methods encompassing the main algorithmic approaches (mutual information, correlation, partial correlation, random forests, support vector machines) using 38 simulated datasets and empirical serous papillary ovarian adenocarcinoma expression-microarray data. We then apply the best-performing method to infer normal and cancer networks. We assess the druggability of the proteins encoded by our predicted target genes using the CancerResource and PharmGKB webtools and databases.
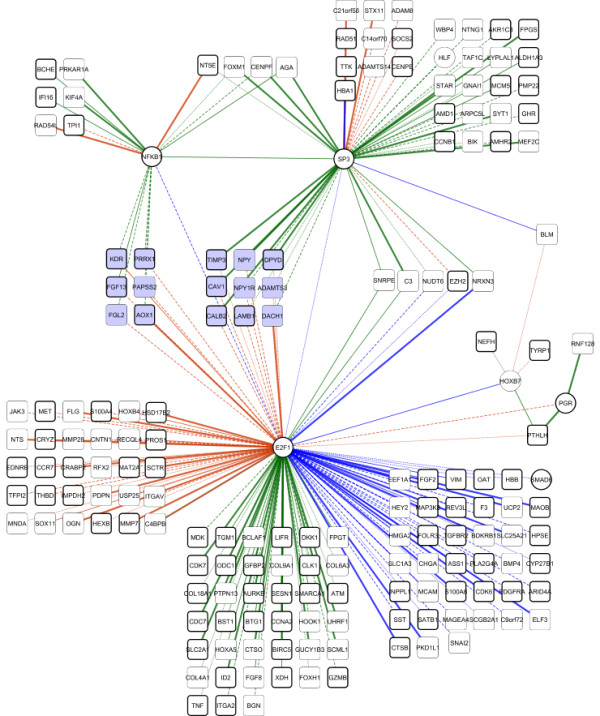
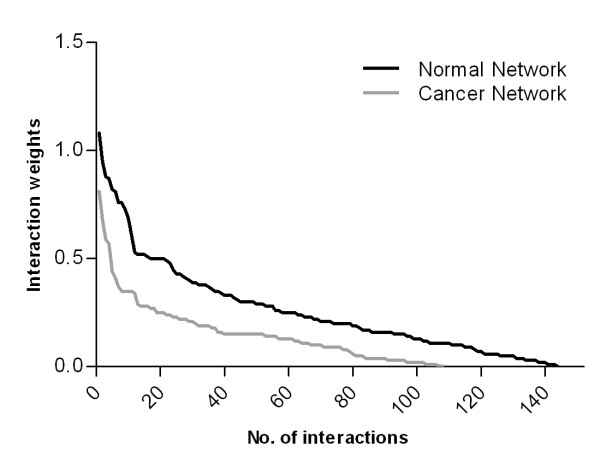
We observe large differences in the accuracy with which these methods predict the underlying gene regulatory network depending on features of the data, network size, topology, experiment type, and parameter settings. Applying the best-performing method (the supervised method SIRENE) to the serous papillary ovarian adenocarcinoma dataset, we infer and rank regulatory interactions, some previously reported and others novel. For selected novel interactions we propose testable mechanistic models linking gene regulation to cancer. Using network analysis and visualization, we uncover cross-regulation of angiogenesis-specific genes through three key transcription factors in normal and cancer conditions. Druggabilty analysis of proteins encoded by the 10 highest-confidence target genes, and by 15 genes with differential regulation in normal and cancer conditions, reveals 75% to be potential drug targets.
Our study represents a concrete application of gene regulatory network inference to ovarian cancer, demonstrating the complete cycle of computational systems biology research, from genome-scale data analysis via network inference, evaluation of methods, to the generation of novel testable hypotheses, their prioritization for experimental validation, and discovery of potential drug targets.
基因调控网络的改变是许多复杂疾病的基础,包括癌症。从高通量微阵列表达数据中推断基因调控网络是计算系统生物学的基本但具有挑战性的任务,并且可以将其转化为基因组医学。尽管已经提出了多种计算和统计方法来解决基因调控网络推断问题,但它们的相对优势和劣势仍然知之甚少,主要是因为比较分析通常只考虑方法的小部分子集,仅使用合成数据,并且/或者未能采用共同的推断质量度量标准。
我们报告了对 9 种最先进的基因调控网络推断方法的全面比较评估,这些方法涵盖了主要的算法方法(互信息、相关、偏相关、随机森林、支持向量机),使用了 38 个模拟数据集和经验性浆液性乳头状卵巢腺癌表达微阵列数据。然后,我们应用表现最佳的方法来推断正常和癌症网络。我们使用 CancerResource 和 PharmGKB 网络工具和数据库评估我们预测的靶基因编码的蛋白质的可药性。
我们观察到这些方法根据数据特征、网络大小、拓扑结构、实验类型和参数设置,在准确预测潜在基因调控网络方面存在很大差异。应用表现最佳的方法(监督方法 SIRENE)对浆液性乳头状卵巢腺癌数据集进行推断,我们推断并对调控相互作用进行排名,其中一些是先前报道的,另一些是新的。对于选定的新型相互作用,我们提出了可测试的将基因调控与癌症联系起来的机制模型。使用网络分析和可视化,我们揭示了正常和癌症条件下血管生成特异性基因通过三个关键转录因子的交叉调控。对 10 个最高置信度靶基因和正常和癌症条件下差异调节的 15 个基因编码的蛋白质进行药物靶标分析,发现 75%的蛋白质可能成为药物靶标。
我们的研究代表了基因调控网络推断在卵巢癌中的具体应用,展示了计算系统生物学研究的完整循环,从基于基因组规模的数据分析到网络推断、方法评估,再到生成新的可测试假设、对其进行优先级排序以进行实验验证以及发现潜在的药物靶标。