School of Computer Science and Engineering, Inha University, Incheon, South Korea.
BMC Bioinformatics. 2012 May 8;13 Suppl 7(Suppl 7):S5. doi: 10.1186/1471-2105-13-S7-S5.
Several computational methods have been developed to predict protein-protein interactions from amino acid sequences, but most of those methods are intended for the interactions within a species rather than for interactions across different species. Methods for predicting interactions between homogeneous proteins are not appropriate for finding those between heterogeneous proteins since they do not distinguish the interactions between proteins of the same species from those of different species.
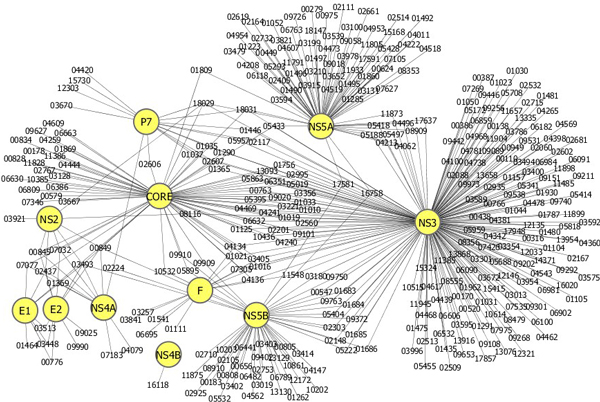
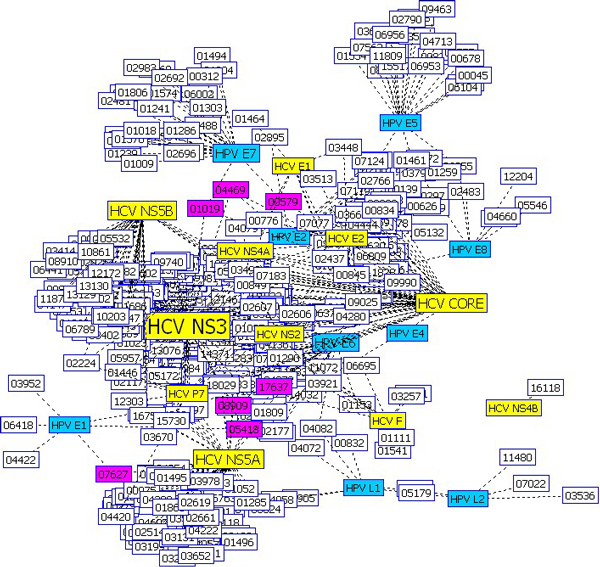
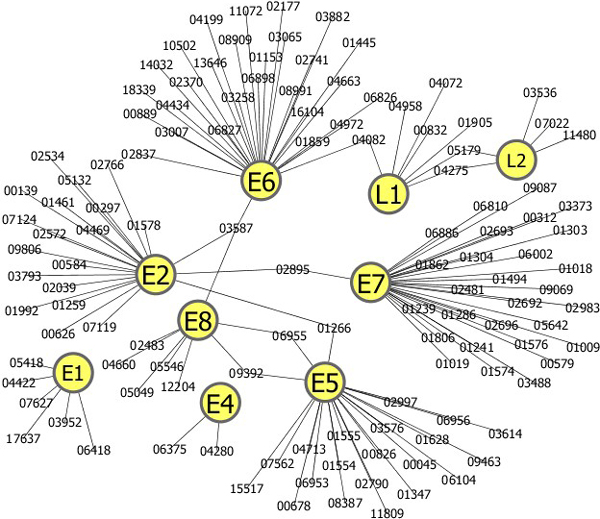
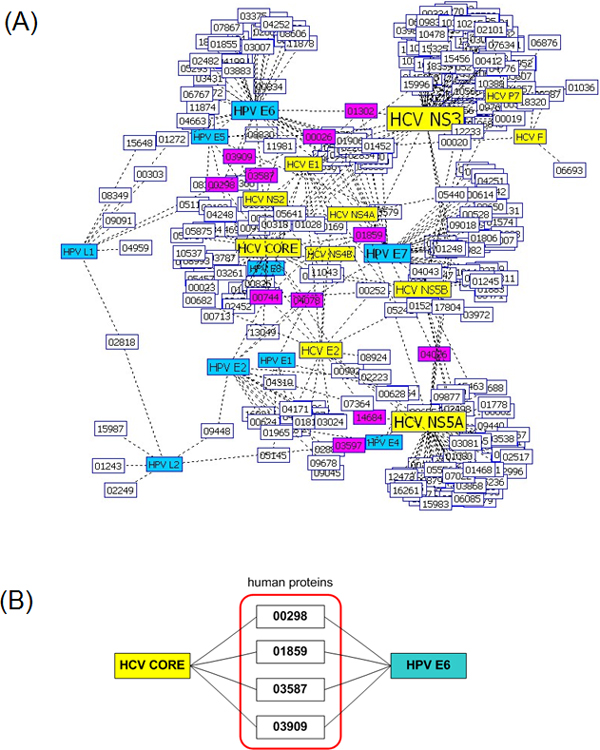
We developed a new method for representing a protein sequence of variable length in a frequency vector of fixed length, which encodes the relative frequency of three consecutive amino acids of a sequence. We built a support vector machine (SVM) model to predict human proteins that interact with virus proteins. In two types of viruses, human papillomaviruses (HPV) and hepatitis C virus (HCV), our SVM model achieved an average accuracy above 80%, which is higher than that of another SVM model with a different representation scheme. Using the SVM model and Gene Ontology (GO) annotations of proteins, we predicted new interactions between virus proteins and human proteins.
Encoding the relative frequency of amino acid triplets of a protein sequence is a simple yet powerful representation method for predicting protein-protein interactions across different species. The representation method has several advantages: (1) it enables a prediction model to achieve a better performance than other representations, (2) it generates feature vectors of fixed length regardless of the sequence length, and (3) the same representation is applicable to different types of proteins.
已经开发出几种从氨基酸序列预测蛋白质-蛋白质相互作用的计算方法,但这些方法大多数旨在预测同一物种内的相互作用,而不是预测不同物种之间的相互作用。用于预测同源蛋白质相互作用的方法不适合寻找异源蛋白质之间的相互作用,因为它们无法区分同一物种蛋白质之间的相互作用与不同物种蛋白质之间的相互作用。
我们开发了一种新方法,用于将可变长度的蛋白质序列表示为固定长度的频率向量,该向量编码序列中三个连续氨基酸的相对频率。我们构建了一个支持向量机 (SVM) 模型来预测与人相互作用的病毒蛋白质。在两种类型的病毒,人乳头瘤病毒 (HPV) 和丙型肝炎病毒 (HCV) 中,我们的 SVM 模型的平均准确率超过 80%,高于具有不同表示方案的另一个 SVM 模型的准确率。使用 SVM 模型和蛋白质的基因本体论 (GO) 注释,我们预测了病毒蛋白和人类蛋白之间的新相互作用。
对蛋白质序列的氨基酸三联体的相对频率进行编码是一种简单而强大的预测不同物种之间蛋白质-蛋白质相互作用的表示方法。该表示方法具有几个优点:(1) 它使预测模型能够实现优于其他表示方法的性能,(2) 它生成固定长度的特征向量,而与序列长度无关,以及 (3) 相同的表示适用于不同类型的蛋白质。