Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, Texas, United States of America.
PLoS Genet. 2012;8(5):e1002713. doi: 10.1371/journal.pgen.1002713. Epub 2012 May 24.
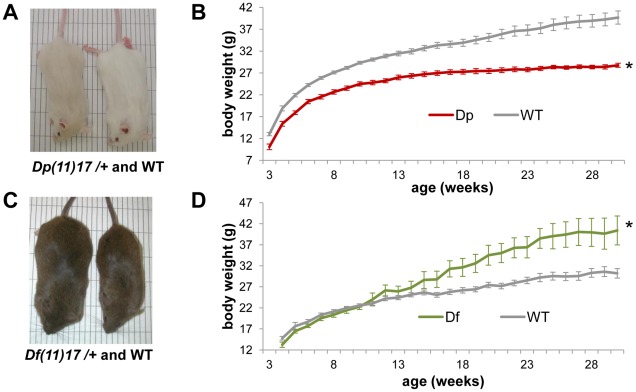
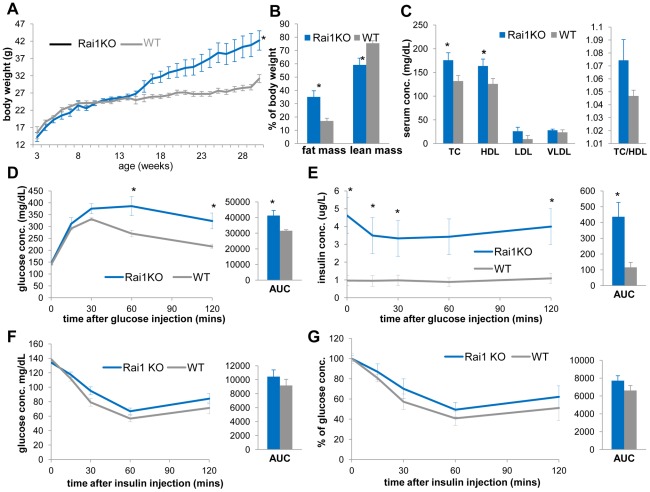
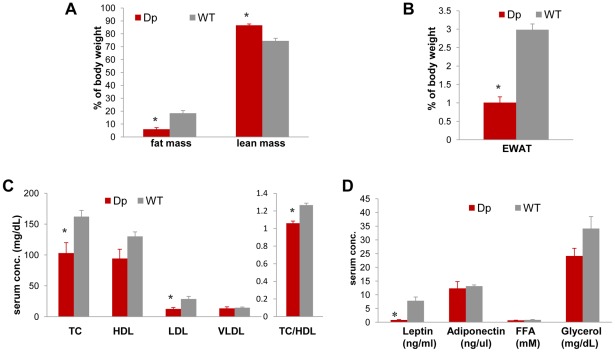
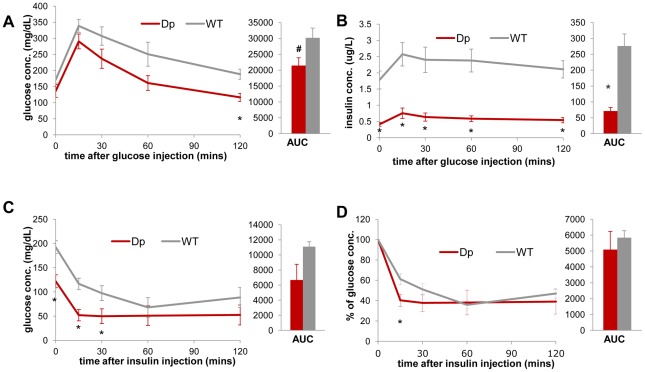
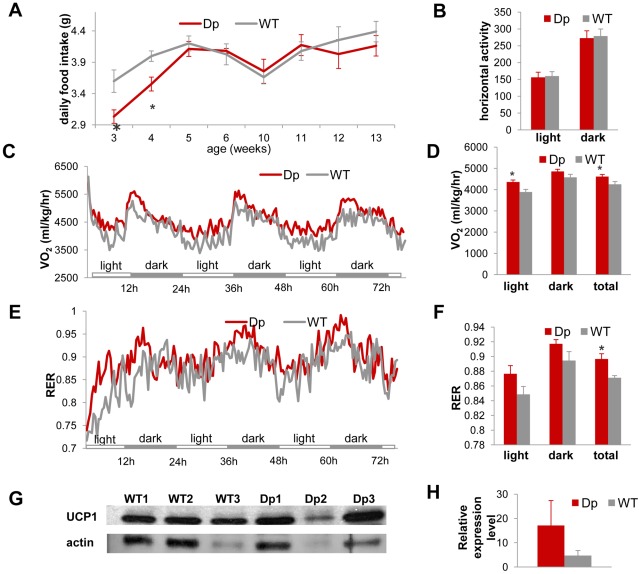
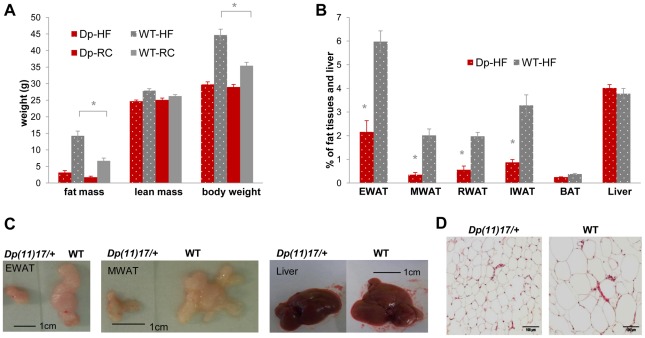
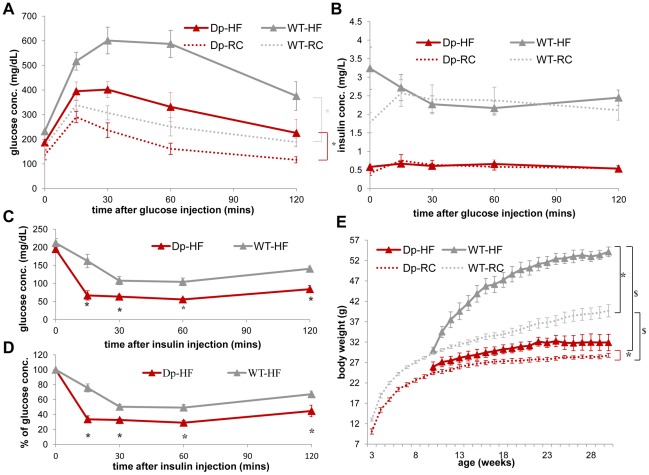
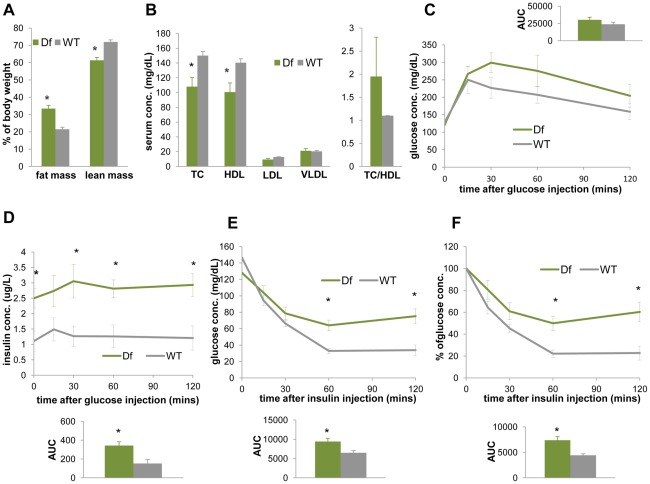
The functional contribution of CNV to human biology and disease pathophysiology has undergone limited exploration. Recent observations in humans indicate a tentative link between CNV and weight regulation. Smith-Magenis syndrome (SMS), manifesting obesity and hypercholesterolemia, results from a deletion CNV at 17p11.2, but is sometimes due to haploinsufficiency of a single gene, RAI1. The reciprocal duplication in 17p11.2 causes Potocki-Lupski syndrome (PTLS). We previously constructed mouse strains with a deletion, Df(11)17, or duplication, Dp(11)17, of the mouse genomic interval syntenic to the SMS/PTLS region. We demonstrate that Dp(11)17 is obesity-opposing; it conveys a highly penetrant, strain-independent phenotype of reduced weight, leaner body composition, lower TC/LDL, and increased insulin sensitivity that is not due to alteration in food intake or activity level. When fed with a high-fat diet, Dp(11)17/+ mice display much less weight gain and metabolic change than WT mice, demonstrating that the Dp(11)17 CNV protects against metabolic syndrome. Reciprocally, Df(11)17/+ mice with the deletion CNV have increased weight, higher fat content, decreased HDL, and reduced insulin sensitivity, manifesting a bona fide metabolic syndrome. These observations in the deficiency animal model are supported by human data from 76 SMS subjects. Further, studies on knockout/transgenic mice showed that the metabolic consequences of Dp(11)17 and Df(11)17 CNVs are not only due to dosage alterations of Rai1, the predominant dosage-sensitive gene for SMS and likely also PTLS. Our experiments in chromosome-engineered mouse CNV models for human genomic disorders demonstrate that a CNV can be causative for weight/metabolic phenotypes. Furthermore, we explored the biology underlying the contribution of CNV to the physiology of weight control and energy metabolism. The high penetrance, strain independence, and resistance to dietary influences associated with the CNVs in this study are features distinct from most SNP-associated metabolic traits and further highlight the potential importance of CNV in the etiology of both obesity and MetS as well as in the protection from these traits.
CNV 对人类生物学和疾病病理生理学的功能贡献还没有得到充分的探索。最近在人类身上的观察结果表明,CNV 与体重调节之间存在初步联系。Smith-Magenis 综合征(SMS)表现为肥胖和高胆固醇血症,是由 17p11.2 上的缺失 CNV 引起的,但有时也由于单个基因 RAI1 的单倍不足引起。17p11.2 的反向重复导致 Potocki-Lupski 综合征(PTLS)。我们之前构建了具有删除,Df(11)17,或重复,Dp(11)17,的小鼠品系,删除或重复的小鼠基因组间隔与 SMS/PTLS 区域同源。我们证明 Dp(11)17 是肥胖的对立面;它传递了一种高度穿透性的、与品系无关的体重减轻、体成分更瘦、TC/LDL 更低和胰岛素敏感性增加的表型,这不是由于食物摄入或活动水平的改变。当喂食高脂肪饮食时,Dp(11)17/+ 小鼠比 WT 小鼠体重增加和代谢变化少得多,表明 Dp(11)17 CNV 可预防代谢综合征。相反,缺失 CNV 的 Df(11)17/+ 小鼠体重增加、脂肪含量更高、HDL 降低和胰岛素敏感性降低,表现出真正的代谢综合征。这些在缺失动物模型中的观察结果得到了来自 76 名 SMS 患者的人类数据的支持。此外,对 knockout/transgenic 小鼠的研究表明,Dp(11)17 和 Df(11)17 CNVs 的代谢后果不仅是由于 SMS 和可能还有 PTLS 的主要剂量敏感基因 Rai1 的剂量改变。我们在人类基因组疾病的染色体工程小鼠 CNV 模型中的实验表明,CNV 可能是体重/代谢表型的原因。此外,我们还探讨了 CNV 对体重控制和能量代谢生理学的贡献的生物学基础。这项研究中的 CNV 具有高穿透性、与品系无关和对饮食影响的抵抗力,这与大多数与 SNP 相关的代谢特征不同,进一步强调了 CNV 在肥胖和代谢综合征的发病机制以及对这些特征的保护中的潜在重要性。