Neural Development Section, Mouse Cancer Genetics Program, Center for Cancer Research, National Cancer Institute, Frederick, Maryland, United States of America.
PLoS One. 2012;7(6):e39946. doi: 10.1371/journal.pone.0039946. Epub 2012 Jun 27.
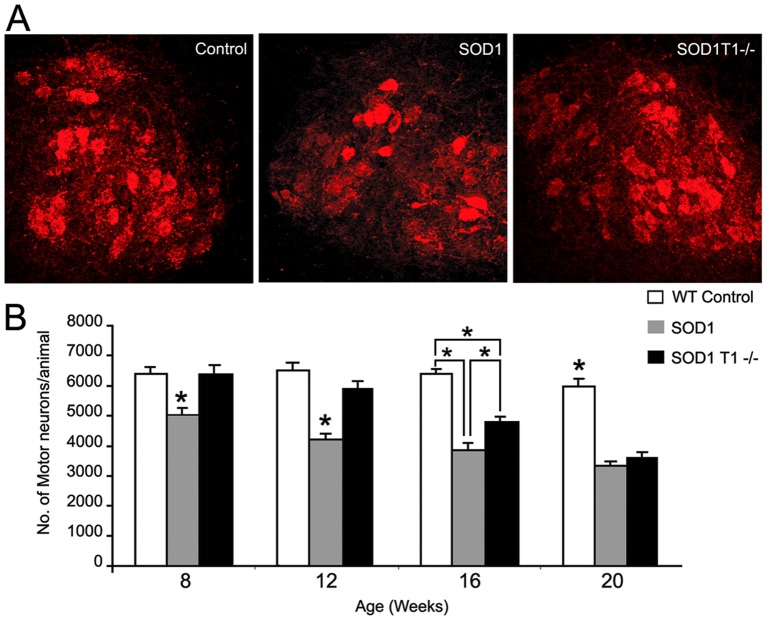
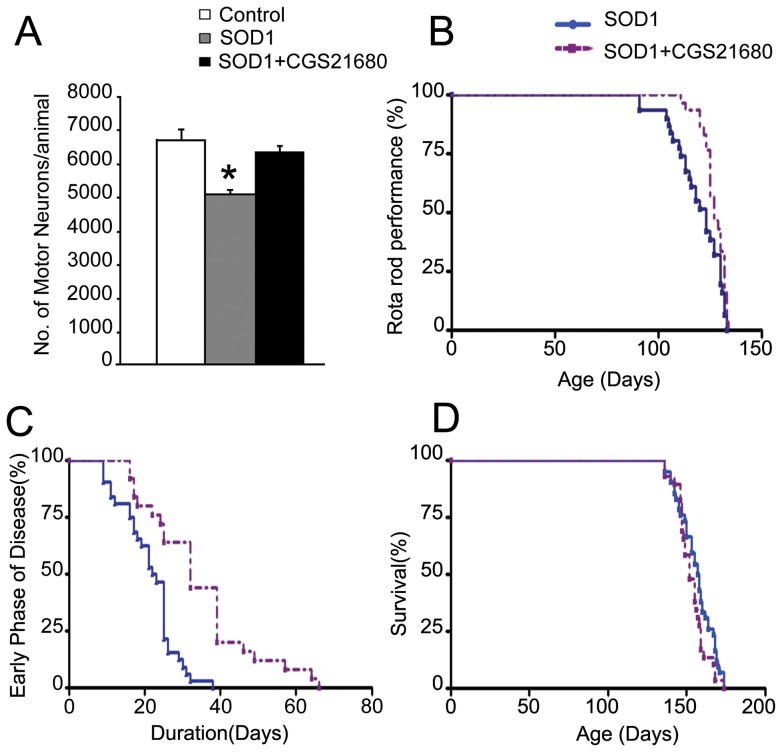
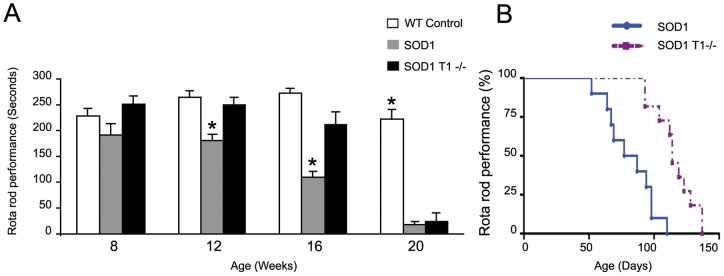
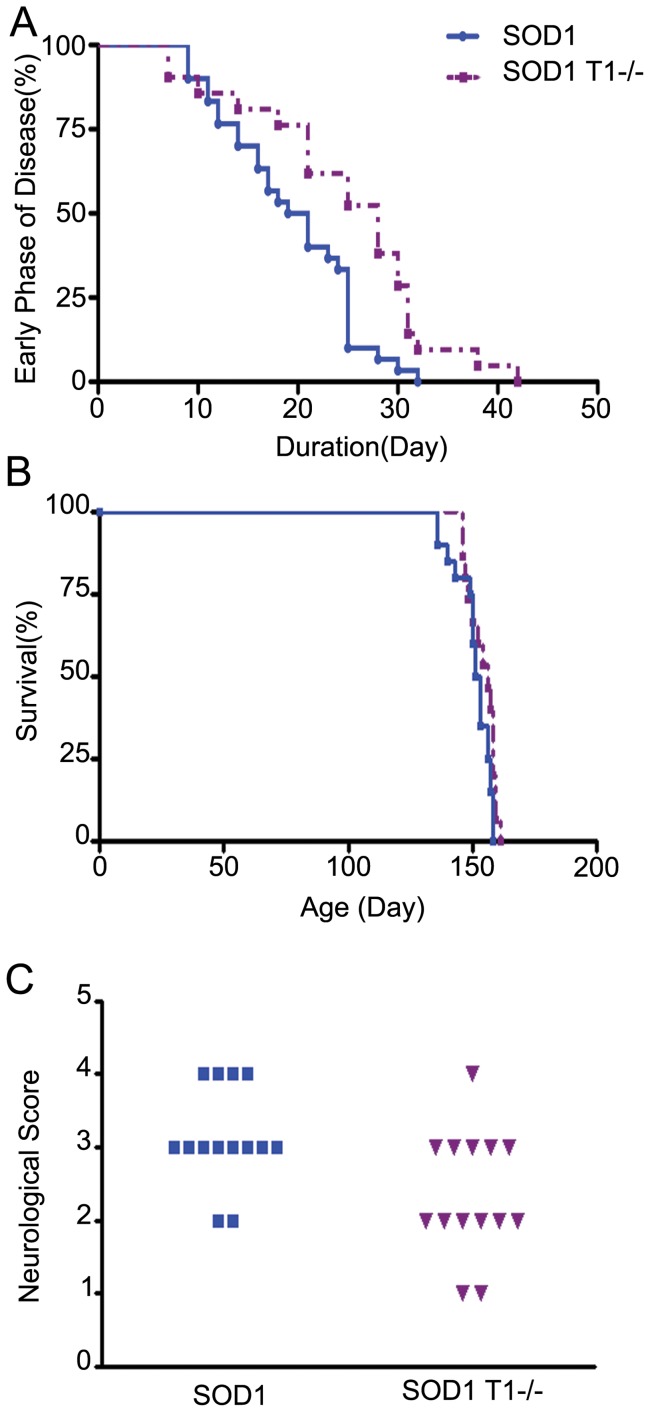
Brain Derived Neurotrophic Factor (BDNF) exerts strong pro-survival effects on developing and injured motoneurons. However, in clinical trials, BDNF has failed to benefit patients with amyotrophic lateral sclerosis (ALS). To date, the cause of this failure remains unclear. Motoneurons express the TrkB kinase receptor but also high levels of the truncated TrkB.T1 receptor isoform. Thus, we investigated whether the presence of this receptor may affect the response of diseased motoneurons to endogenous BDNF. We deleted TrkB.T1 in the hSOD1(G93A) ALS mouse model and evaluated the impact of this mutation on motoneuron death, muscle weakness and disease progression. We found that TrkB.T1 deletion significantly slowed the onset of motor neuron degeneration. Moreover, it delayed the development of muscle weakness by 33 days. Although the life span of the animals was not affected we observed an overall improvement in the neurological score at the late stage of the disease. To investigate the effectiveness of strategies aimed at bypassing the TrkB.T1 limit to BDNF signaling we treated SOD1 mutant mice with the adenosine A2A receptor agonist CGS21680, which can activate motoneuron TrkB receptor signaling independent of neurotrophins. We found that CGS21680 treatment slowed the onset of motor neuron degeneration and muscle weakness similarly to TrkB.T1 removal. Together, our data provide evidence that endogenous TrkB.T1 limits motoneuron responsiveness to BDNF in vivo and suggest that new strategies such as Trk receptor transactivation may be used for therapeutic intervention in ALS or other neurodegenerative disorders.
脑源性神经营养因子(BDNF)对发育中和受损的运动神经元发挥强大的生存促进作用。然而,在临床试验中,BDNF 未能使肌萎缩侧索硬化症(ALS)患者受益。迄今为止,这种失败的原因仍不清楚。运动神经元表达 TrkB 激酶受体,但也高水平表达截断的 TrkB.T1 受体同工型。因此,我们研究了这种受体的存在是否会影响患病运动神经元对内源性 BDNF 的反应。我们在 hSOD1(G93A)ALS 小鼠模型中删除了 TrkB.T1,并评估了这种突变对运动神经元死亡、肌肉无力和疾病进展的影响。我们发现,TrkB.T1 缺失显著减缓了运动神经元退化的发生。此外,它使肌肉无力的发展延迟了 33 天。尽管动物的寿命没有受到影响,但我们观察到在疾病后期神经评分的整体改善。为了研究旨在绕过 TrkB.T1 限制 BDNF 信号的策略的有效性,我们用腺苷 A2A 受体激动剂 CGS21680 治疗 SOD1 突变小鼠,CGS21680 可以激活运动神经元 TrkB 受体信号,而无需神经营养因子。我们发现,CGS21680 治疗可减缓运动神经元退化和肌肉无力的发生,与 TrkB.T1 缺失相似。总之,我们的数据提供了证据,表明内源性 TrkB.T1 限制了运动神经元对体内 BDNF 的反应性,并表明新的策略,如 Trk 受体转激活,可用于 ALS 或其他神经退行性疾病的治疗干预。