Burnett School of Biomedical Sciences, College of Medicine, University of Central Florida, Orlando, FL 32816, USA.
Neurobiol Dis. 2013 Mar;51:72-81. doi: 10.1016/j.nbd.2012.07.004. Epub 2012 Jul 20.
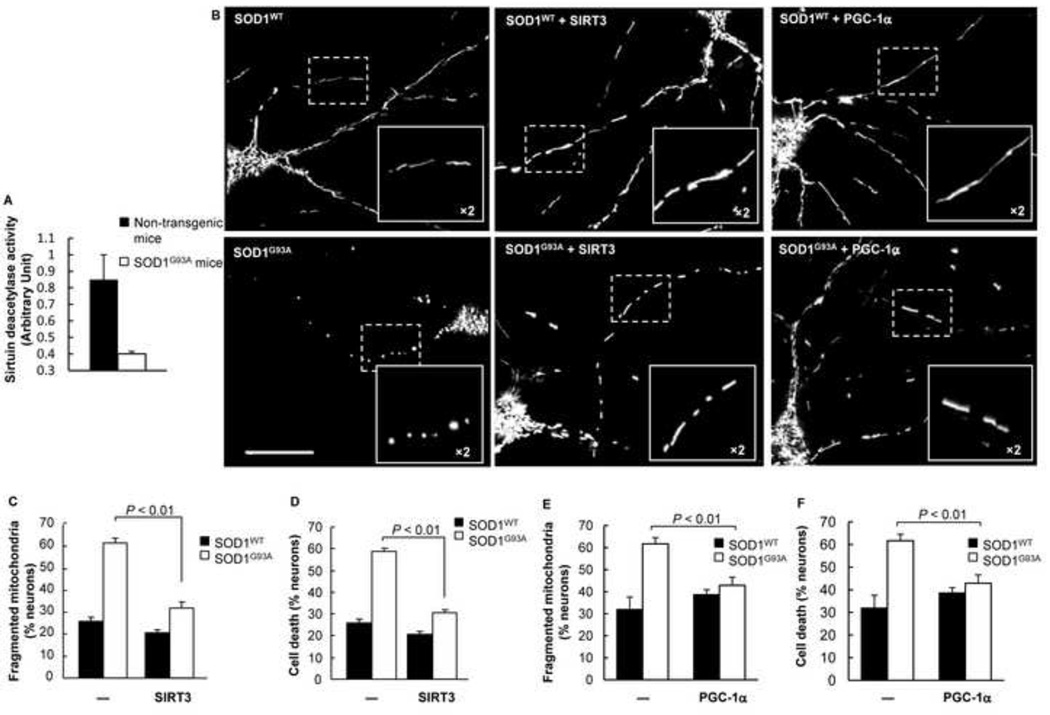
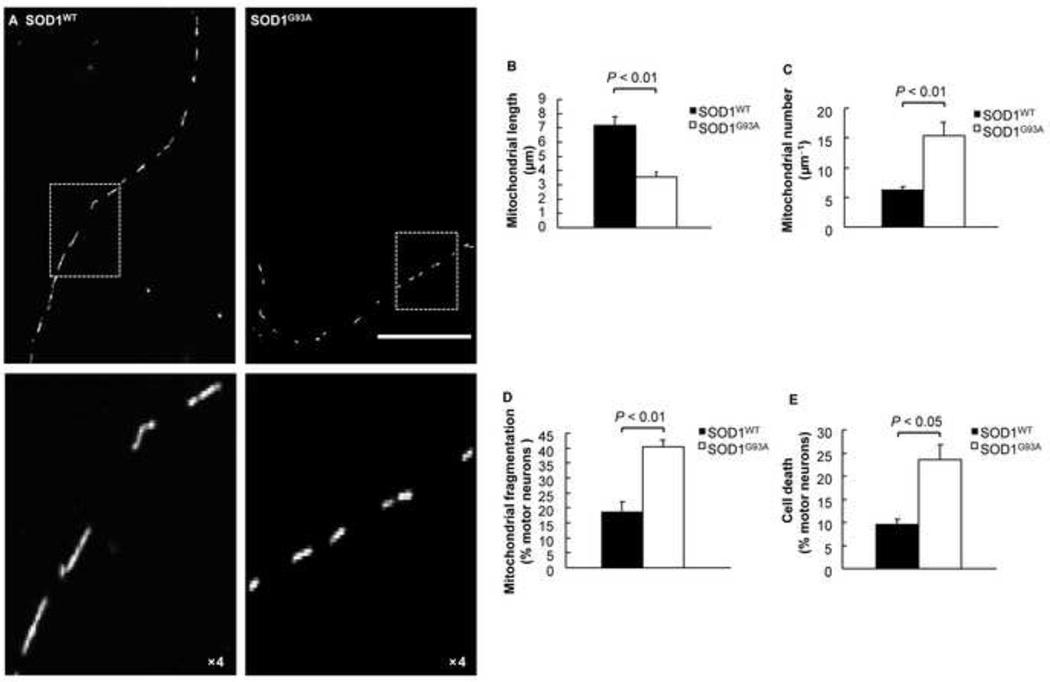
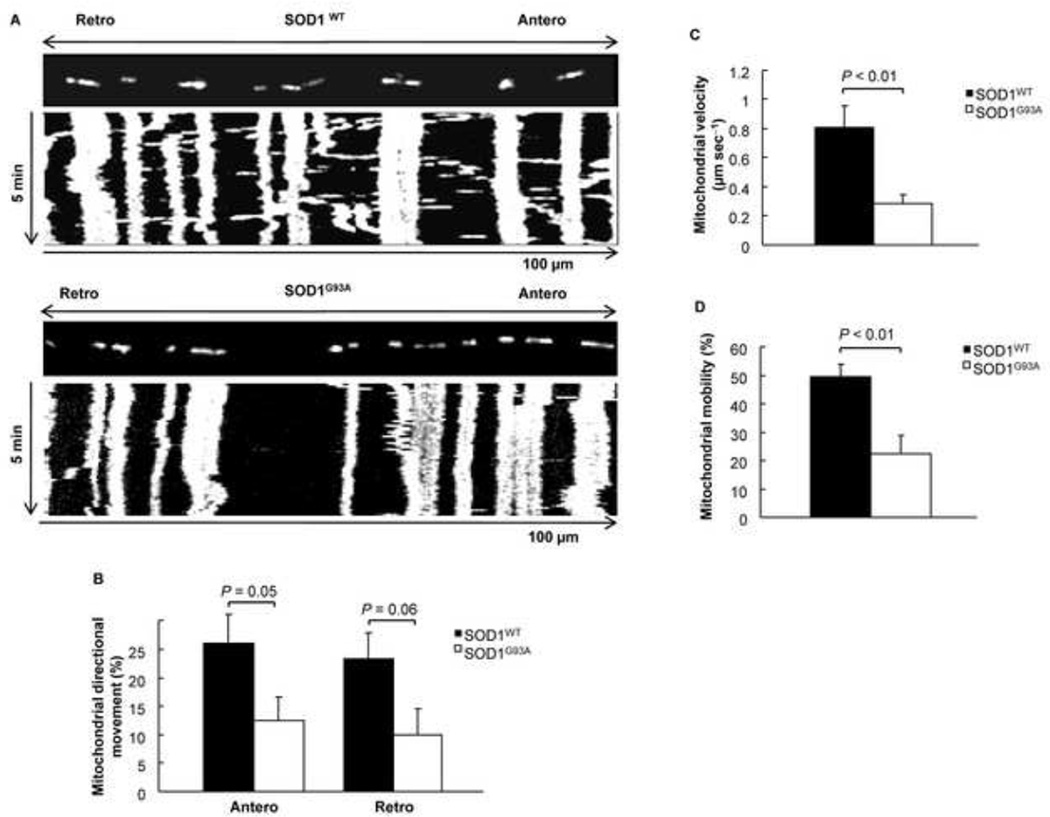
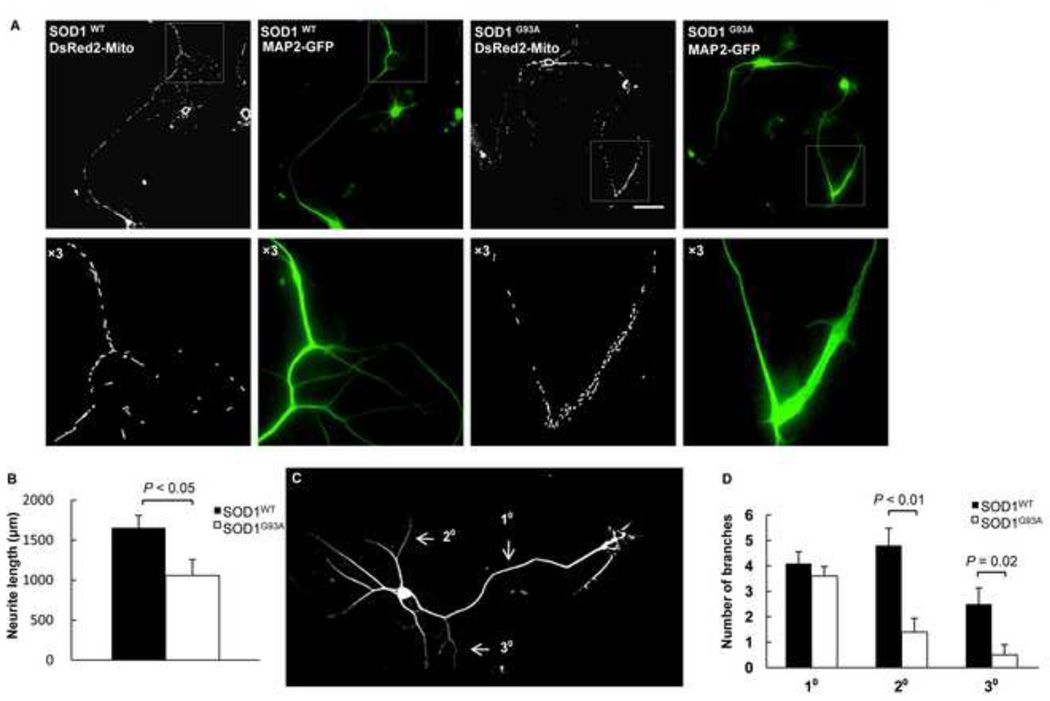
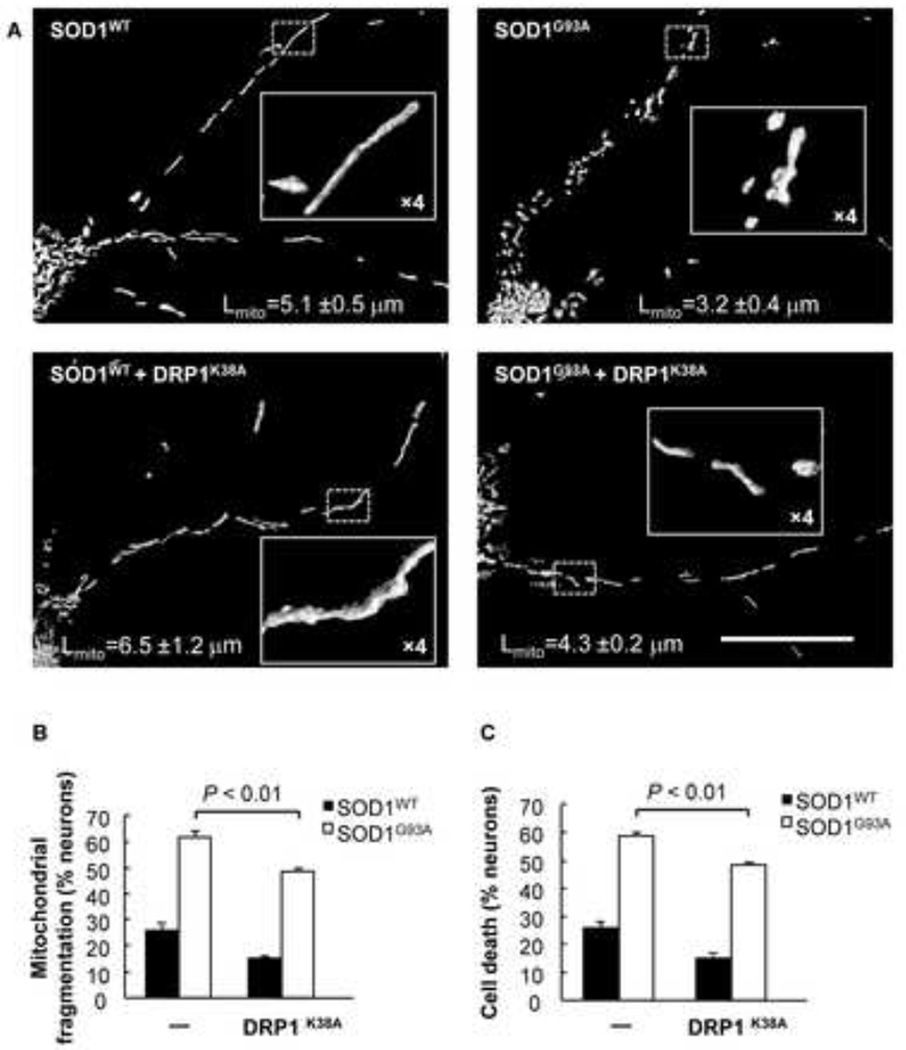
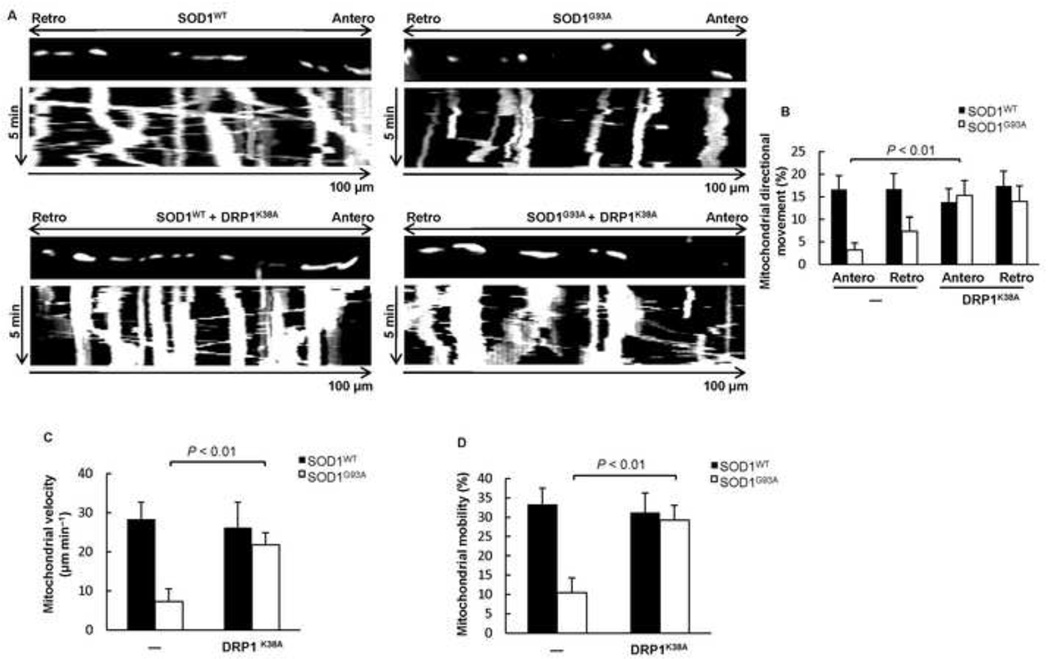
Mutations in the Cu/Zn Superoxide Dismutase (SOD1) gene cause an inherited form of ALS with upper and lower motor neuron loss. The mechanism underlying mutant SOD1-mediated motor neuron degeneration remains unclear. While defects in mitochondrial dynamics contribute to neurodegeneration, including ALS, previous reports remain conflicted. Here, we report an improved technique to isolate, transfect, and culture rat spinal cord motor neurons. Using this improved system, we demonstrate that mutant SOD1(G93A) triggers a significant decrease in mitochondrial length and an accumulation of round fragmented mitochondria. The increase of fragmented mitochondria coincides with an arrest in both anterograde and retrograde axonal transport and increased cell death. In addition, mutant SOD1(G93A) induces a reduction in neurite length and branching that is accompanied with an abnormal accumulation of round mitochondria in growth cones. Furthermore, restoration of the mitochondrial fission and fusion balance by dominant-negative dynamin-related protein 1 (DRP1) expression rescues the mutant SOD1(G93A)-induced defects in mitochondrial morphology, dynamics, and cell viability. Interestingly, both SIRT3 and PGC-1α protect against mitochondrial fragmentation and neuronal cell death by mutant SOD1(G93A). This data suggests that impairment in mitochondrial dynamics participates in ALS and restoring this defect might provide protection against mutant SOD1(G93A)-induced neuronal injury.
铜/锌超氧化物歧化酶(SOD1)基因的突变导致具有上下运动神经元丧失的遗传性 ALS。突变 SOD1 介导的运动神经元变性的机制尚不清楚。虽然线粒体动力学的缺陷导致神经退行性变,包括 ALS,但以前的报告仍然存在冲突。在这里,我们报告了一种改进的技术,用于分离、转染和培养大鼠脊髓运动神经元。使用这个改进的系统,我们证明突变 SOD1(G93A) 导致线粒体长度显著缩短和圆形片段化线粒体的积累。碎片化线粒体的增加与顺行和逆行轴突运输的阻滞以及细胞死亡的增加同时发生。此外,突变 SOD1(G93A) 诱导轴突伸长和分支减少,伴随着生长锥中圆形线粒体的异常积累。此外,通过表达显性失活的动力相关蛋白 1 (DRP1) 恢复线粒体裂变和融合平衡,可挽救突变 SOD1(G93A) 诱导的线粒体形态、动力学和细胞活力缺陷。有趣的是,SIRT3 和 PGC-1α 均可通过突变 SOD1(G93A) 防止线粒体碎片化和神经元细胞死亡。该数据表明,线粒体动力学的损伤参与了 ALS,并且恢复这种缺陷可能为对抗突变 SOD1(G93A)诱导的神经元损伤提供保护。