Department of Biomedical Informatics, Vanderbilt University School of Medicine, Nashville, Tennessee, United States of America.
PLoS One. 2012;7(7):e41292. doi: 10.1371/journal.pone.0041292. Epub 2012 Jul 23.
Several studies have reported gene expression signatures that predict recurrence risk in stage II and III colorectal cancer (CRC) patients with minimal gene membership overlap and undefined biological relevance. The goal of this study was to investigate biological themes underlying these signatures, to infer genes of potential mechanistic importance to the CRC recurrence phenotype and to test whether accurate prognostic models can be developed using mechanistically important genes.
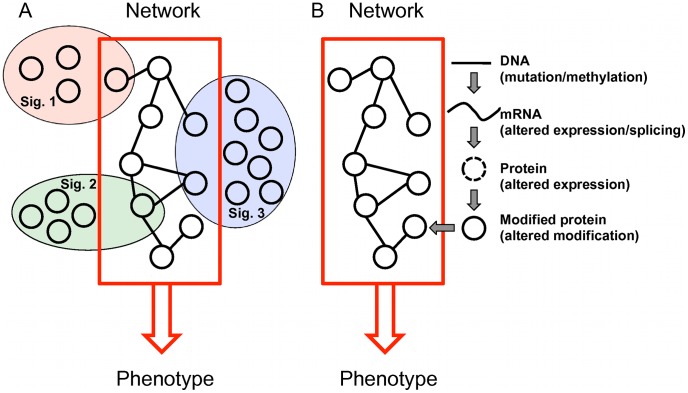
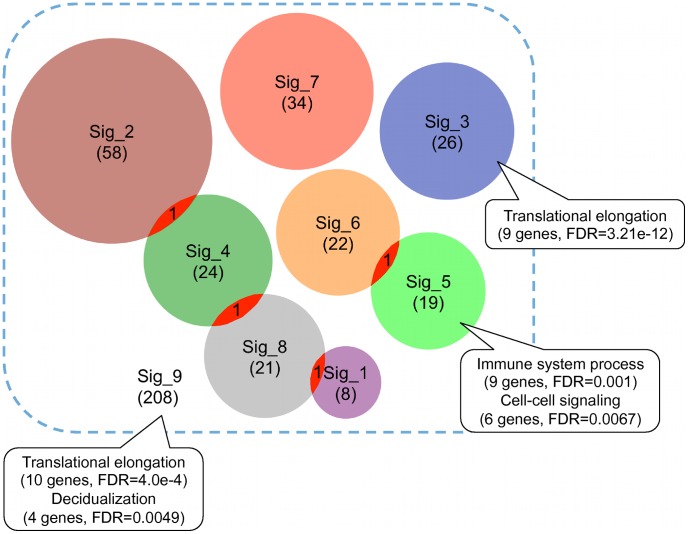
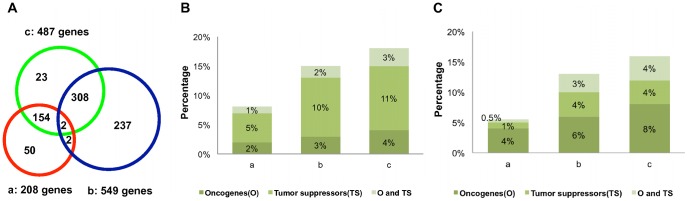
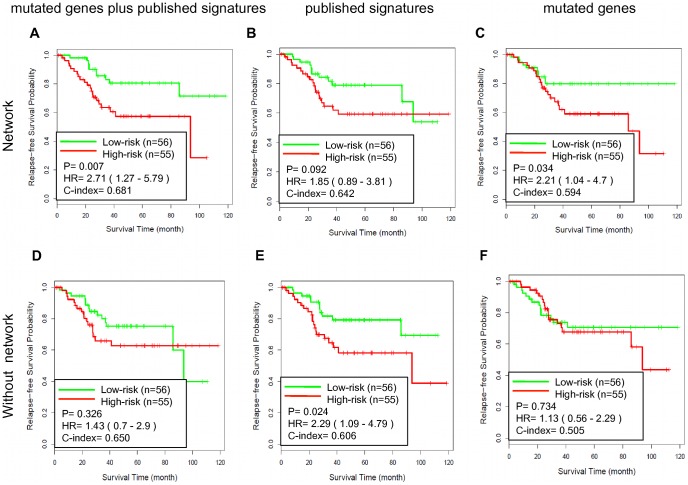
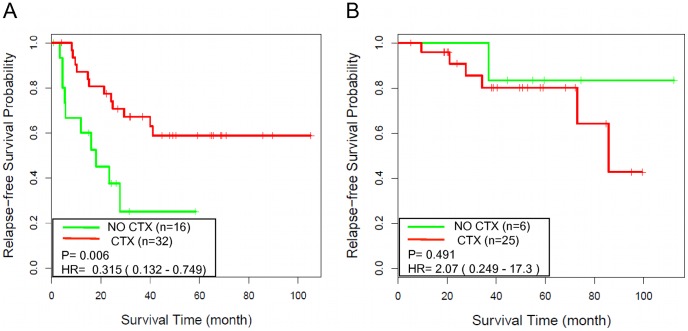
We investigated eight published CRC gene expression signatures and found no functional convergence in Gene Ontology enrichment analysis. Using a random walk-based approach, we integrated these signatures and publicly available somatic mutation data on a protein-protein interaction network and inferred 487 genes that were plausible candidate molecular underpinnings for the CRC recurrence phenotype. We named the list of 487 genes a NEM signature because it integrated information from Network, Expression, and Mutation. The signature showed significant enrichment in four biological processes closely related to cancer pathophysiology and provided good coverage of known oncogenes, tumor suppressors, and CRC-related signaling pathways. A NEM signature-based Survival Support Vector Machine prognostic model was trained using a microarray gene expression dataset and tested on an independent dataset. The model-based scores showed a 75.7% concordance with the real survival data and separated patients into two groups with significantly different relapse-free survival (p = 0.002). Similar results were obtained with reversed training and testing datasets (p = 0.007). Furthermore, adjuvant chemotherapy was significantly associated with prolonged survival of the high-risk patients (p = 0.006), but not beneficial to the low-risk patients (p = 0.491).
The NEM signature not only reflects CRC biology but also informs patient prognosis and treatment response. Thus, the network-based data integration method provides a convergence between biological relevance and clinical usefulness in gene signature development.
有几项研究报告了基因表达特征,这些特征可预测 II 期和 III 期结直肠癌(CRC)患者的复发风险,这些特征的基因成员重叠最小,且生物学相关性尚未确定。本研究的目的是研究这些特征背后的生物学主题,推断对 CRC 复发表型具有潜在机制重要性的基因,并测试是否可以使用具有机制重要性的基因来开发准确的预后模型。
我们研究了八个已发表的 CRC 基因表达特征,在基因本体论富集分析中没有发现功能上的收敛。我们使用基于随机游走的方法,整合了这些特征和公开的体细胞突变数据在蛋白质-蛋白质相互作用网络上,并推断出 487 个可能是 CRC 复发表型潜在分子基础的基因。我们将这 487 个基因的列表命名为 NEM 特征,因为它整合了来自网络、表达和突变的信息。该特征在与癌症病理生理学密切相关的四个生物学过程中表现出显著的富集,并很好地涵盖了已知的癌基因、肿瘤抑制基因和 CRC 相关信号通路。使用微阵列基因表达数据集训练基于 NEM 特征的生存支持向量机预后模型,并在独立数据集上进行测试。基于模型的评分与真实生存数据的一致性达到 75.7%,并将患者分为两组,无复发生存差异显著(p=0.002)。使用反转的训练和测试数据集也得到了类似的结果(p=0.007)。此外,辅助化疗与高危患者的生存延长显著相关(p=0.006),但对低危患者无益处(p=0.491)。
NEM 特征不仅反映了 CRC 生物学,还为患者预后和治疗反应提供了信息。因此,基于网络的数据分析方法为基因特征开发中的生物学相关性和临床实用性提供了融合。