Department of Biostatistics, Vanderbilt University Medical Center, Nashville, Tennessee, United States of America.
Center for Quantitative Sciences, Vanderbilt University Medical Center, Nashville, Tennessee, United States of America.
PLoS Biol. 2018 Oct 22;16(10):e2006687. doi: 10.1371/journal.pbio.2006687. eCollection 2018 Oct.
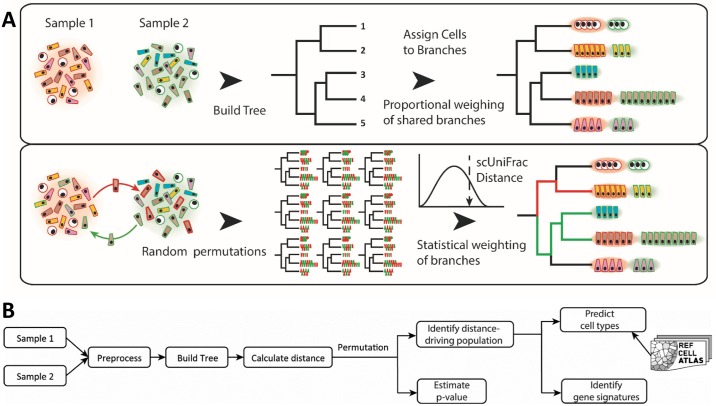
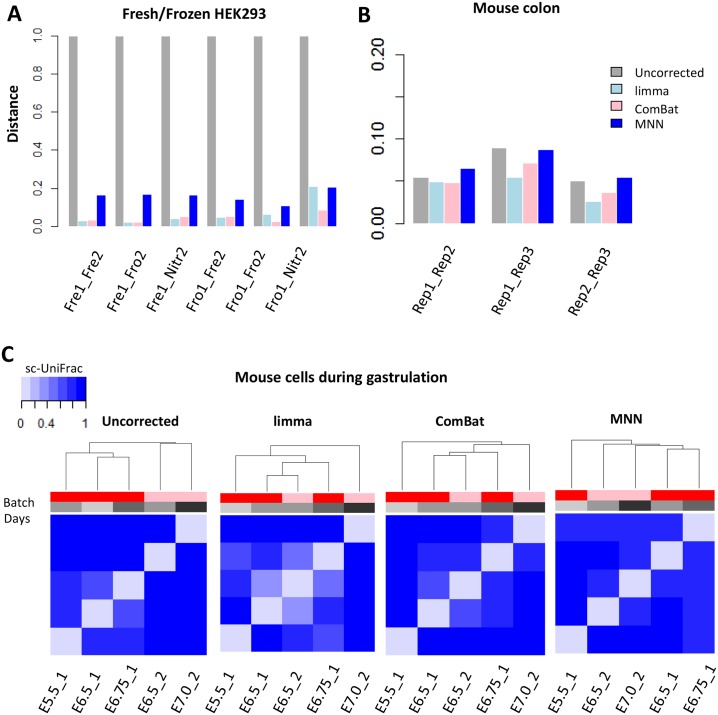
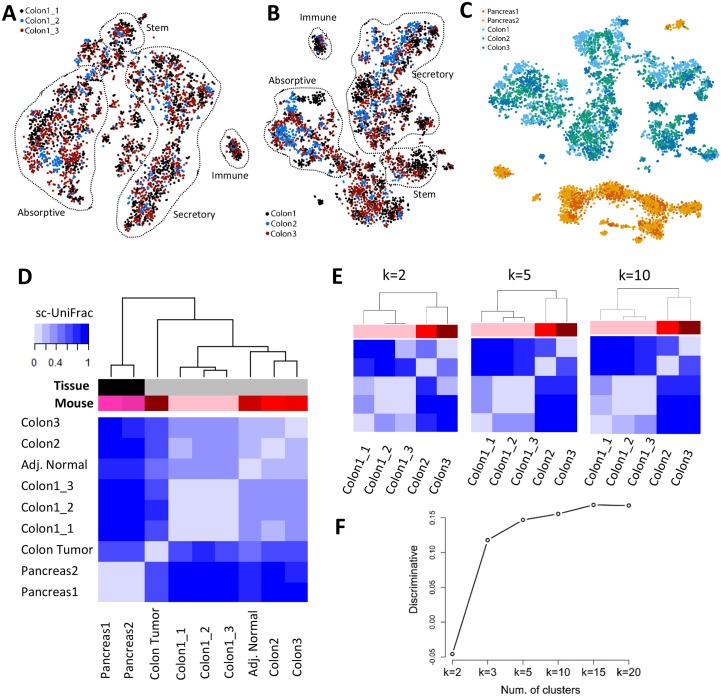
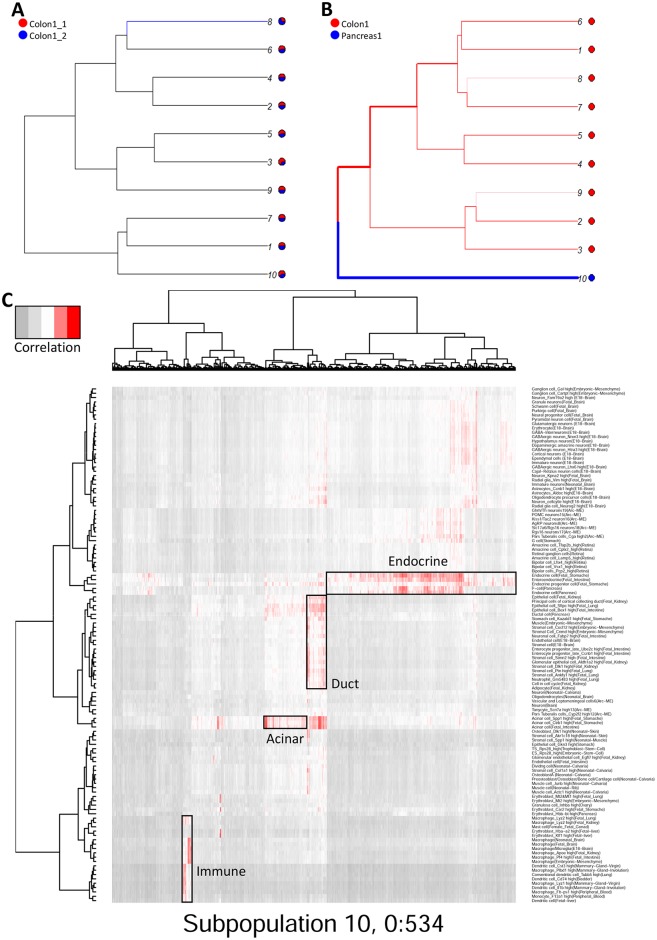
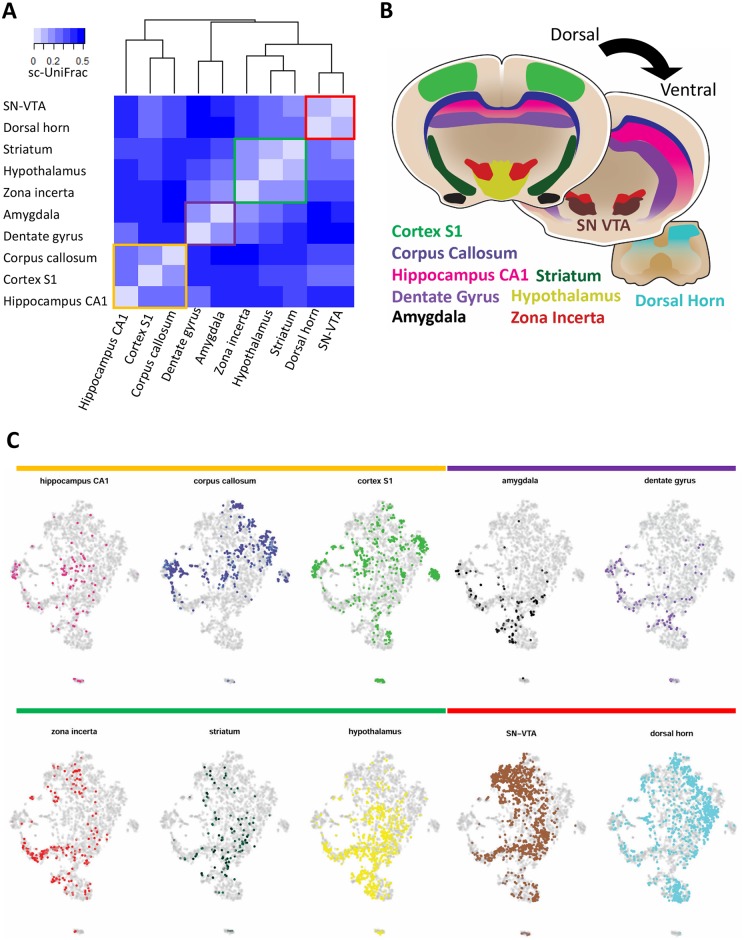
Single-cell RNA sequencing (scRNA-seq) has become a powerful tool for the systematic investigation of cellular diversity. As a number of computational tools have been developed to identify and visualize cell populations within a single scRNA-seq dataset, there is a need for methods to quantitatively and statistically define proportional shifts in cell population structures across datasets, such as expansion or shrinkage or emergence or disappearance of cell populations. Here we present sc-UniFrac, a framework to statistically quantify compositional diversity in cell populations between single-cell transcriptome landscapes. sc-UniFrac enables sensitive and robust quantification in simulated and experimental datasets in terms of both population identity and quantity. We have demonstrated the utility of sc-UniFrac in multiple applications, including assessment of biological and technical replicates, classification of tissue phenotypes and regional specification, identification and definition of altered cell infiltrates in tumorigenesis, and benchmarking batch-correction tools. sc-UniFrac provides a framework for quantifying diversity or alterations in cell populations across conditions and has broad utility for gaining insight into tissue-level perturbations at the single-cell resolution.
单细胞 RNA 测序 (scRNA-seq) 已成为系统研究细胞多样性的有力工具。随着许多计算工具的发展,用于识别和可视化单个 scRNA-seq 数据集中的细胞群体,因此需要有方法来定量和统计定义细胞群体结构在数据集之间的比例变化,例如细胞群体的扩张或收缩、出现或消失。这里我们提出了 sc-UniFrac,这是一个用于在单细胞转录组景观之间的细胞群体中统计量化组成多样性的框架。sc-UniFrac 能够在模拟和实验数据集中以群体身份和数量的角度进行敏感和稳健的量化。我们已经在多个应用中证明了 sc-UniFrac 的实用性,包括评估生物学和技术重复、组织表型和区域特化的分类、鉴定和定义肿瘤发生中的改变的细胞浸润、以及基准批处理校正工具。sc-UniFrac 提供了一种用于量化细胞群体在不同条件下的多样性或变化的框架,对于深入了解单细胞分辨率下的组织水平扰动具有广泛的应用价值。