Department of Bioinformatics and Genomics, University of North Carolina at Charlotte, Charlotte, North Carolina, United States of America.
PLoS One. 2012;7(7):e41294. doi: 10.1371/journal.pone.0041294. Epub 2012 Jul 26.
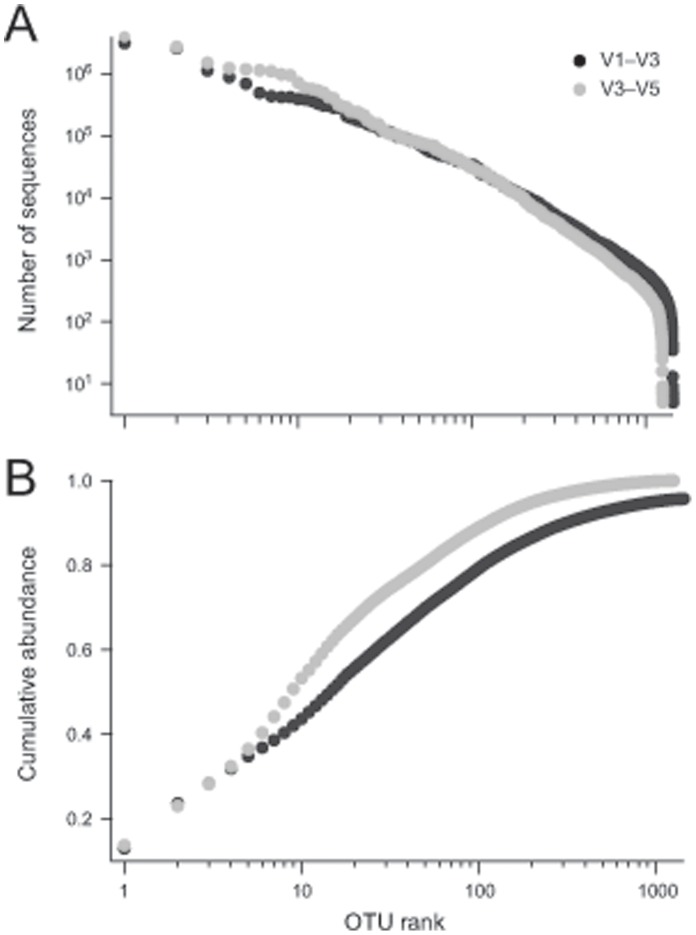
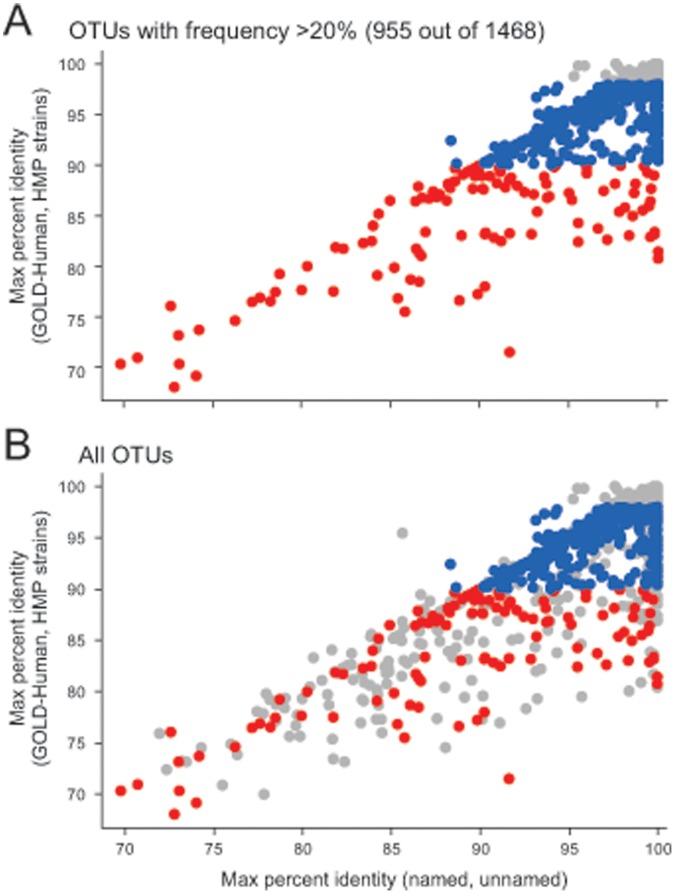
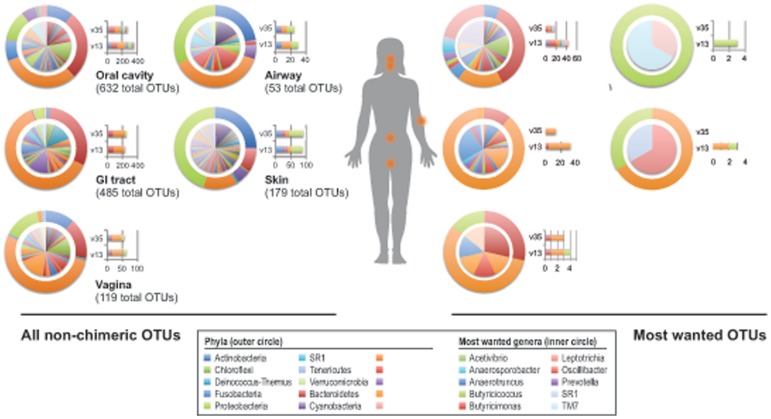
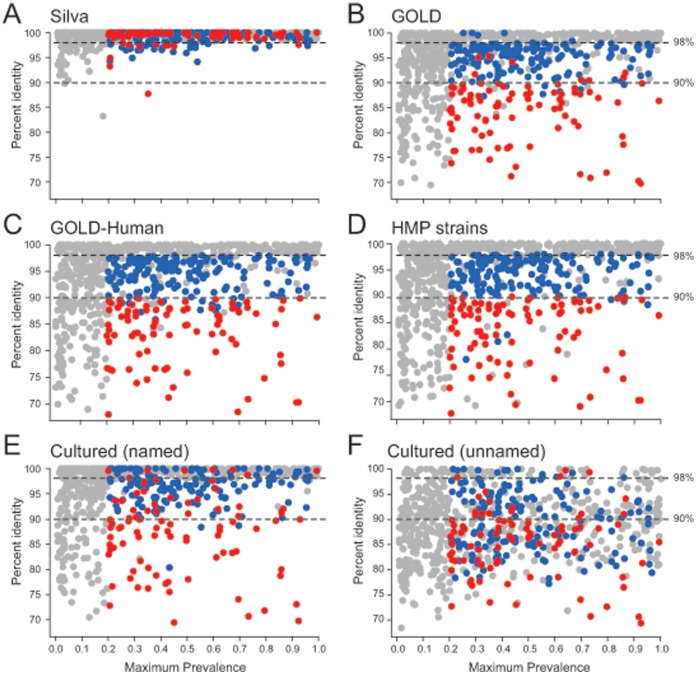
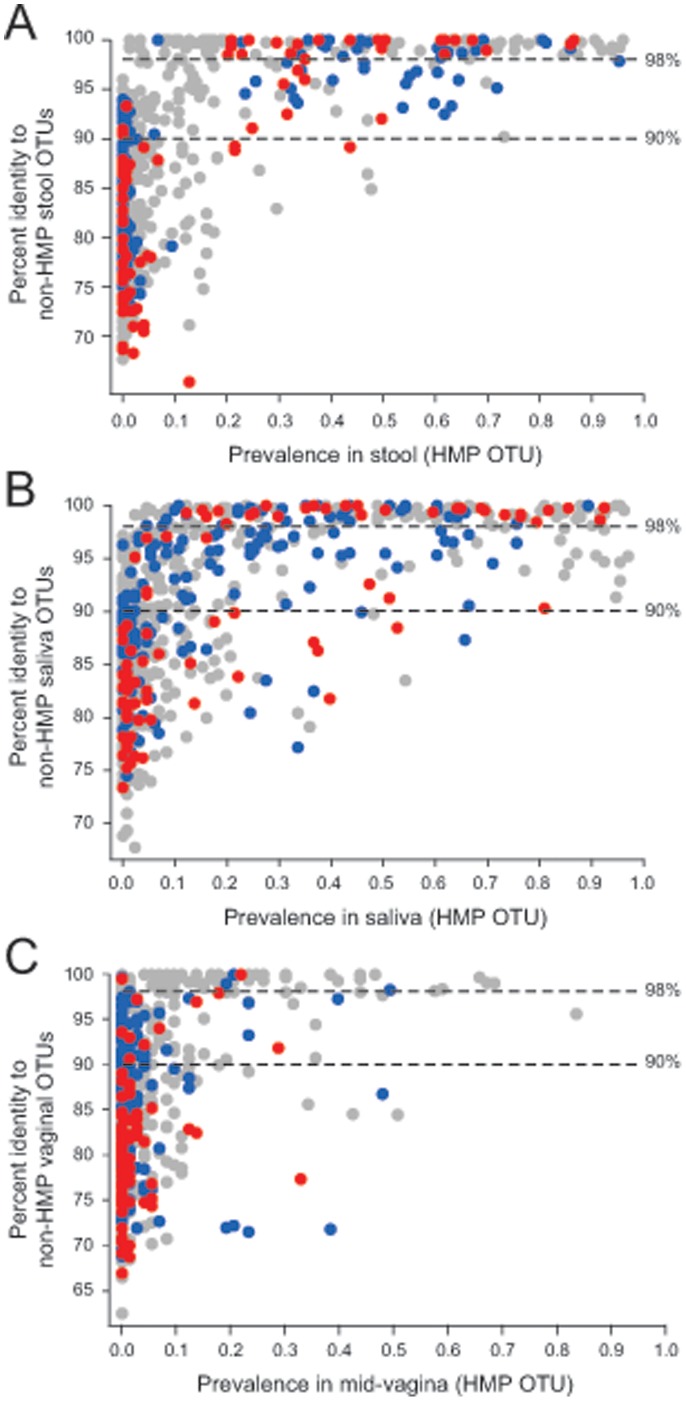
The goal of the Human Microbiome Project (HMP) is to generate a comprehensive catalog of human-associated microorganisms including reference genomes representing the most common species. Toward this goal, the HMP has characterized the microbial communities at 18 body habitats in a cohort of over 200 healthy volunteers using 16S rRNA gene (16S) sequencing and has generated nearly 1,000 reference genomes from human-associated microorganisms. To determine how well current reference genome collections capture the diversity observed among the healthy microbiome and to guide isolation and future sequencing of microbiome members, we compared the HMP's 16S data sets to several reference 16S collections to create a 'most wanted' list of taxa for sequencing. Our analysis revealed that the diversity of commonly occurring taxa within the HMP cohort microbiome is relatively modest, few novel taxa are represented by these OTUs and many common taxa among HMP volunteers recur across different populations of healthy humans. Taken together, these results suggest that it should be possible to perform whole-genome sequencing on a large fraction of the human microbiome, including the 'most wanted', and that these sequences should serve to support microbiome studies across multiple cohorts. Also, in stark contrast to other taxa, the 'most wanted' organisms are poorly represented among culture collections suggesting that novel culture- and single-cell-based methods will be required to isolate these organisms for sequencing.
人类微生物组计划(HMP)的目标是生成一份全面的人类相关微生物目录,其中包括代表最常见物种的参考基因组。为此,HMP 已经使用 16S rRNA 基因(16S)测序技术对 200 多名健康志愿者的 18 个身体栖息地的微生物群落进行了特征描述,并从人类相关微生物中生成了近 1000 个参考基因组。为了确定当前的参考基因组集合在多大程度上捕获了健康微生物组中的多样性,并指导微生物组成员的分离和未来测序,我们将 HMP 的 16S 数据集与几个参考 16S 集合进行了比较,以创建一个用于测序的“最需要”的分类群列表。我们的分析表明,HMP 队列微生物组中常见分类群的多样性相对适中,这些 OTU 代表的新分类群较少,许多 HMP 志愿者共有的常见分类群在不同健康人群中反复出现。总之,这些结果表明,应该有可能对人类微生物组的很大一部分进行全基因组测序,包括“最需要”的部分,并且这些序列应该能够支持多个队列的微生物组研究。此外,与其他分类群形成鲜明对比的是,“最需要”的生物在培养物集中的代表性很差,这表明需要新的培养物和单细胞为基础的方法来分离这些用于测序的生物。